附: QT间期延长综合征
出处:按学科分类—医药、卫生 上海科学技术文献出版社《临床心律失常诊疗手册》第66页(1855字)
长QT综合征(LQTS)为基因突变导致心肌膜离子通道功能障碍致心室复极延长的一组症候群,病死率极高。有症状而未接受治疗的病例其15年内死亡率超过60%,有效的治疗可使死亡率下降至3%~4%。该综合征有先天性及获得性两种,获得性者多为抗心律失常药物或电解质紊乱所引起。先天性长QT综合征,又称特发性长QT综合征,首例系由Jervell-Lange-Nielsen(1957年)所报道,表现为先天性耳聋、晕厥、恶性心律失常、猝死,亦称维尔朗厄·尼尔逊(Jervell-Lange-Nielsen)综合征,至1963年及1964年,Romano和Ward分别报道另一组特发性长QT综合征,特点相同,但不伴耳聋称Romano-Ward综合征。本例病程长达8年,晕厥反复发生,可除外药源性且不伴耳聋,故符合Romano-Ward综合征诊断。自1991年以来针对此综合征的深入研究表明,本病为隐性基因遗传的分子疾病,涉及多个离子流的复杂病理改变。目前已确定有4种基因突变分别位于11,7,3,21号染色体上,另有报道于4号染色体上的第5种基因突变。LQT1为KVLQT1基因突变影响心肌细胞的Iks。LQT2为HERG基因突变致钾通道异常影响Ikr。LQT3为ScN5A基因突变致钠通道异常。LQT4为mink(KCNE1)基因突变。上述类型几乎含括了近半数LQTS患者的病变原因。有认为,症状与基因类型有关,LQT1与LQT5症状发作90%于运动和情绪激动时;LQT390%于睡眠中;而LQT2几乎均见于运动、激动、熟睡与唤醒之间。
有关LQTS的治疗,每需依其发病机制而定。现认为起搏治疗可缩短由ScN5A缺陷所致的QT延长,而LQT1、LQT2心律失常发生多与交感神经兴奋、儿茶酚胺释放有关,故治疗针对调节心脏自主神经的功能,包括应用β受体阻滞剂;此外,LQT1,2尚与钾离子外流有关,故此类患者的治疗除给予β受体阻滞剂外,若增用钾通道开放剂,其疗效将更为明显。
鉴于近年来,LQTS发病机制在分子生物学水平上有了新的认识,故选择针对基因缺陷所致离子通道的新药定将有助于进一步提高本病治疗的成功率。
上海市静安区中心医院曾报道长QT综合征并尖端扭转型室速1例,经紧急心脏临时起搏,超速抑制,并配伍静脉输注氯化钾及口服较大剂量普萘洛尔(160mg/d)后病情稳定,好转出院。
患者女性,14岁,反复晕厥8年,再发加重2小时,患者于6岁时,无明显诱因出现反复发作晕厥,意识丧失,历时短暂,持续约半分钟可自然恢复,无抽搐及二便失禁,至7岁起发作更见频繁,每周约1~2次,多伴抽搐,每需作胸外按压后苏醒。9岁时于体育课跑步时出现晕厥,继而心搏骤停,后经心肺复苏,心脏临时起搏器以及人工呼吸机等在内历时3周的抢救后获救。尔后长期服用普萘洛尔(心得安)(120mg/d),6年内未再发作,无耳聋,否认家族史。入院前1周患者曾有“感冒”病史,入院前2小时情绪激动后突发晕厥,随即家人予胸外心脏按压后,神志转清。片刻后再次发作,急送本院急诊,心电图示:QT间期延长(>0.60s),频发多源室早,阵发性尖端扭转型室速(附图1),乃急行心脏临时起搏,超速抑制(起搏频率120bpm)后收心脏监护病房。实验室检查:血钾3.5mmol/L,血镁0.9mmol/L(0.67~1.0mmol/L),CKMB 19U/L(0~24U/L)。进入心脏监护病房后,虽患者可在频率120bpm的起搏心律控制下,病情获改善,但期间胸闷不适症状仍时有发作,且每伴频发成串期前收缩,稍降低调搏频率即可加剧异位节律的出现。结合患者焦虑并血钾处正常低值,遂增加静脉输注氯化钾的用量(4g/d),并使普萘洛尔(心得安)加量至160mg/d,历时1周许,血钾测值维持在4.0mmol/L,撤去起搏器,病情稳定,窦性心律75次/分,QT 0.48s,无室性期前收缩(附图2),好转出院。

附图1 QT延长>0.06s,RonT室早,尖端扭转型室速
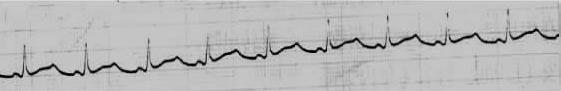
附图2 恢复窦性心律。QT(QU)仍达0.48s(QTc0.53s)