先天性胆管闭锁
出处:按学科分类—医药、卫生 科学技术文献出版社《肝胆胰外科疾病诊断标准》第212页(2016字)
一、概述
先天性胆管闭锁(congential biliary atresia)是先天性发育障碍所致的胆管梗塞。
二、流行病学
先天性胆管闭锁是新生儿期间梗阻性黄疸的常见原因。病变可累及整个胆管,也可仅累及肝内或肝外的部分胆管,其中以肝外胆管闭锁常见,占85%~90%。发病率女性高于男性。
三、病因
本病的病因至今尚未完全了解,目前主要有两种学说。
1.先天性发育畸形学说 胚胎早期,原始胆管已形成,后为增生的上皮细胞填塞,随后上皮细胞发生空泡化,并相互融合贯通而形成胆管系统。若胚胎期2~3个月时发育障碍,胆管无空泡化或空泡化不完全,则形成胆管全部或部分闭锁。胆管闭锁常合并下腔静脉缺如,门静脉异位等畸形。有人认为,本病与染色体异常有关。
2.病毒感染学说 近来的研究认为,胆管闭锁可能是获得性疾病。胚胎后期或出生早期患者病毒感染,可引起胆管上皮毁损,胆管周围炎及纤维性变等而引起胆管部分或完全闭锁。还有人认为胆管闭锁与新生儿肝炎是同一疾病过程中不同时期的表现,肝炎可波及肝外胆管引起胆管及胆管周围组织发生炎性改变而导致胆管闭锁。乙型肝炎发病率较高的东南亚国家,胆管闭锁发病也较高。
四、生理病理
胆管先天性发育畸形大多为胆管闭锁,仅极少数呈狭窄改变。胆管闭锁所致梗阻性黄疸,可致肝细胞损害,肝因淤胆而显着肿大、变硬,呈暗绿色或褐绿色,肝功能异常。若胆管梗阻不能及时解除,则可发展为胆汁性肝硬化,晚期为不可逆性改变。按闭锁部位可分为三型:
(1)肝内型:占大多数,可为肝内大胆管,或肝内小胆管,或全部胆管。
(2)肝外型:可发生在肝外胆管任何部位,但肝内胆管正常。
(3)混合型:肝内外胆管全部闭锁。
五、临床表现
1.黄疸 梗阻性黄疸是本病突出表现。一般出生时并无黄疸,1~2周后出现,呈进行性加深。巩膜褐色、皮肤由金黄变为绿褐色或暗绿色,大便渐为白陶土色,尿色随黄疸加深而呈浓茶样,尿布染黄。皮肤有瘙痒抓痕。2~3个月后可发生出血倾向及凝血机制障碍。
2.营养及发育不良 初期患儿情况良好,营养发育正常,表现与黄疸深度不相符。随后一般情况逐渐恶化,至3~4个月时出现营养不良、贫血、发育迟缓、反应迟钝等。
3.肝脾肿大 是本病特点。出生时肝脏正常,随病情发展而呈进行性肿大,3个月左右即可发展为胆汁性肝硬化及门静脉高血压症。最终常因感染、出血、肝衰竭、肝昏迷,于出生后1年内死亡。
六、诊断与诊断标准
凡出生后1~2个月出现持续性黄疸,白陶土色大便,伴肝肿大者均应怀疑本病。下列各点有助于确诊:
(1)黄疸超过3~4周仍呈进行性加重,对利胆药物治疗无效;对苯巴比妥和激素治疗试验无反应;血清胆红素呈持续上升。
(2)十二指肠引流液内无胆汁。
(3)B超检查显示肝外胆管和胆囊发育不良或缺如。
(4)99mTc—EHIDA扫描肠内无核素显示。
七、鉴别诊断
本病需与新生儿胆汁浓缩相鉴别,后者常见于新生儿肝炎、溶血病、药物(维生素K)核严重脱水等引起胆汁浓缩、排出不畅而致暂时性梗阻性黄疸。一般经1~2个月利胆或激素治疗后黄疸逐渐减轻至消退。B超检查对鉴别诊断有帮助。
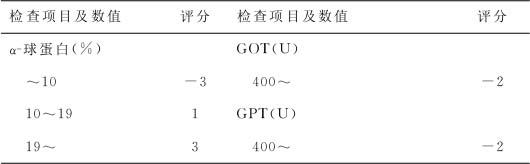
日本葛西森夫推荐的胆管闭锁及新生儿肝炎鉴别评分见表20—1。
表20—1 胆管闭锁及新生儿肝炎鉴别评分表