射线探测器
出处:按学科分类—工业技术 企业管理出版社《计量专业工程师手册》第517页(13698字)
所有射线探测器的原理都基于第10.2.3节中提到的射线与物质的相互作用,人们应用这些相互作用原理,制造出了各种类型的探测器。这里着重介绍经常使用的几类探测器。
1.气体探测器
气体探测器包括电离室、正比计数管和盖革-米勒计数管三种类型,它们收集射线在探测器气体中电离产生的电荷,使其成为可记录的信号。使气体分子电离产生一个电子和一个离子(称为离子对)所需的能量称为电离功,一般用W表示,也称W值。因此射线在气体中损失能量至少要大于W值才有被记录的可能,几种气体的W值列在表10.2-1中。电离产生的离子和电子可能相遇复合为中性分子,电子也可能被分子吸附成为负离子,它们都是作无规则运动,与正离子相遇复合,因此不能被收集成为可记录的信号。在加有电场空间则是另外一种情况(见图10.2-4),电子向附加电场的正极运动,离子则向负极运动,在回路中形成电流,从而在负载两端(ab两点产生电压(电流),提供可能被记录的信号。随着工作电压V0的改变,在不同能量的入射粒子情况下,a.b两点的电压有不同的变化。
表10.2-1 不同气体的W值


图10.2-4 气体探测器电流回路示意路
图10.2-5示出了某入射粒子能量为2MeV和1MeV时变化情况。图中A表示ab两点的输出电压(用对数坐标),V是附加工作电压。在O和V1之间,A随V增加,在此区间内能量高的粒子产生的电压也较高。在V1-V2区间内A不随V变化,但是2MeV的入射粒子引起的输出电压几乎是1MeV的入射粒子的两倍,这一区间就是电离室工作的区间,称为电离室区。工作电压继续上升超过V2时,输出电压也增加,直至V3的这段区间内,输出电压与入射粒子的能量成正比,所以称这个区间为正比区。若工作电压继续上升直至V4,输出电压仍随互作电压增加,并仍可以区分不同能量的入射粒子,但不再与入射粒子的能量成正比,这个区间称为有限正比区。在工作电压超过V4的情况下,输出电压已与入射粒子的能量无关,即无论入射粒子的能量多少,只要它与计数管气体起作用,它的输出电压是相同的,这个区域称为盖革(Geiger)区。
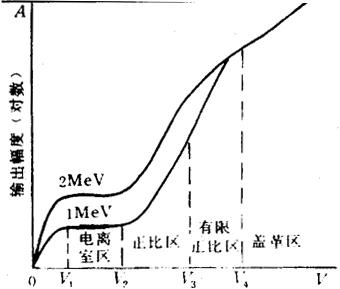
图10.2-5 气体探测器输出信号幅度与附加电压的关系
(1)电离室
当气体计数管在电离室区域工作时,电离产生的电子与离子不再产生复合,电荷全部被收集,而且这一区域的电场强度还不够高到足以使初级电离产生的电子产生次级电离,因此输出信号(电流或脉冲幅度)只与初级电离产生的电荷有关。电离室分电流型脉冲型两种工作方式,当辐射在电离室中产生的电离事件频率较高,在一个电离事件以后的电荷收集时间内又有更多的粒子进入电离室产生新的电离,使得电离室无法区分各个事件,这时使用电流型电离室,测量辐射在单位时间内产生的平均电流。脉冲型电离室则用于较弱的辐射,测量单个电离事件形成的脉冲。在电流电离室情况下,入射粒子在电离室气体中产生电离,如果辐射场稳定,在附加电压情况下,电流在开始时随电压上升(不同的辐射场电流也不同),升至一定电压后,电流不再随电压上升,即进入饱和区。在饱和区内,在电离室有效体积内形成的电离电荷全被收集,因此原则上测量的电流反映了电离室有效体积内由于射线电离形成的电荷。在脉冲电离室情况下,电离室中单个粒子在电离室中产生的电离事件可以以脉冲输出方式被记录,它不仅可以测量粒子数,还可以测量粒子的能量,因此一定程度上也可用于射线的能谱分析。电离室的工作电阻R的两端可有一脉冲输出,最大幅度可达:
V=N0e/C
式中 N0为射线一个粒子产生的初始离子对数,e为电子电荷,C为极间电容。
用于能量分析应采用很大的RC常数,而用于计数率的测量应采用较小的RC常数。
为了适应不同的使用目的,电离室有圆柱形、平板形式或井形等。
(2)正比计数管。气体计数管工作在正比区时,初始电离产生的离子对不仅没有复合,而且由于电场较强,所产生的电子又可产生多级次级电离形成离子雪崩。以柱状的计数管为例,一般阳极为位于中心的金属丝,阴极为金属壁,若设它们的半径分别为a及b,则距计数管轴中心处r(a<r<b)处的电场强度为Er=V0/(rln(b/a),其中V0为加在计数管两极的电压。由于在阳极丝附近可以产生电离雪崩,一个初始电离产生的这种电离雪崩现象称为气体放大,若初始电离产生一个电子,经气体放大后平均电子数为m,则称m为气体放大系数。在正比区内,m不随初始电离的离子对的数目改变,输出电压与初始电离产生的电荷保持正比关系。由于气体放大的存在,即使初始电离只产生一个离子对,正比计数管也可能获得可以处理的信号。为了获得合适的气体放大倍数,计数管中所充的气体必须没有明显的吸附电子的效应。正比计数管都是以脉冲方式工作,它不仅可用于测量射线在气体中发生气体事件的数目,而且可用于分析能量。这里和脉冲电离室的情况一样,用于能量分析时要求较大RC常数,用于计数测量则需较小的RC常数。
(3)盖革-米勒(Geiger-Mtiller)计数管。它通常称为盖革管或GM管。当电场强度增加时,初始电子与气体碰撞时产生的光子可在电极上产生数量大于1的光电子,这种次级光电子又可重复上述效应,这时输出脉冲的幅度已与初始离子对数目无关。因此一个初始离子对就可给出足够大的输出脉冲。如果不采用“淬灭”(quenching)措施,盖革计数管可能自续放电或出现倍加信号脉冲。加淬灭的方法有两种,一种方法是加淬灭电子线路,使得一旦输出端有脉冲输出,电路就给出反馈信号,使得GM计数管极间电压下降,从而减少了阳极附近的电场强度,不再有放电现象产生;另一种方法是计数管中加淬灭气体,这类气体(例如酒精)能被电子电离,但是它不会产生光电效应,因此也不会产生自续放电现象。
2.闪烁计数器
有许多物质在射线的作用下能产生用肉眼可见的荧光。光电倍增管的发明,使得用荧光测量射线的方法,即闪烁计数方法重新得到发展并被广泛使用。
理想的闪烁材料应具有以下特点:入射的射线的能量转换成可见光的效率高;吸收的能量与发射的光产额有很好的线性关系;材料对于本身发射的光是透明的;发光的衰减时间短;易于加工成合适的形状及足够大的体积;对光的反射率应与光电倍增管的玻璃接近以便于配合。但是没有任何一种材料能同时具有以上几种特点,因此必须根据自己的使用目的和实验条件来选择合适的闪烁体,闪烁材料可分为有机和无机两种,它们产生荧光的机制不相同。一般来说,无机材料光输出量高,但反应时间慢,而有机材料光输出量少,但反应时间快,由于无机材料的原子序数较高,适合γ射线探测和能谱分析,而有机材料通常适合于β-射线和快中子。
(1)有机闪烁体
有机材料中的荧光过程产生于单个分子能级之间的变化,因此对于某给定的分子形式,观察荧光时与它的物理状态无关,即不管它是固态、液态或气态。
①有机闪烁晶体。只有两种有机晶体得到较广泛应用,即蒽晶体(Anthracene)和芪晶体(Stilbene),前者在所有有机闪烁体中发光效率最高,后者发光效率较低,但它适用于波形甄别以区别不同带电粒子和β粒子引起的闪烁。这两种晶体都较脆,不易加工成大的尺寸,而且闪烁效率与射线入射的方向相对于晶体的角度有关。
②液体闪烁体。它由具有闪烁特性的溶质和合适的溶剂组成。溶质可分为两种,分别称为第一溶质(第一闪烁剂)和第二溶质(第二闪烁剂)。这是因为第一溶质吸收射线的能量以后发出的荧光不一定能与光电倍增管的波长响应匹配,第二溶质起着波长转换作用,它将第一溶质发的光吸收,然后发出能与光电倍增管响应匹配的荧光,所以第二溶质又称波长转换剂或波长漂移剂。常用的溶剂有甲苯、对二甲苯和二氧杂环乙烷,第一溶质有PPO,丁基PBD和PBD等,第二溶质有PO PO P,M 2-P OP OP等。
③其它有机闪烁体。可将有机闪烁剂溶于溶剂中,然后聚合成固体塑料,得到塑料闪烁体,它可加工成不同的形状;也可将有机闪烁体制成薄膜,厚度约为20μg/cm,因此它可做测量质子和α-粒子的穿透型探测器。如果在有机闪烁体中加入少量高原子序物质,例如铅,就成为含铅有机闪烁体,它提高了测γ-射线光电峰的效率,可用于测γ-射线。
(2)无机闪烁体。
卤化盐闪烁体。这一类的闪烁体有Nal(T1)、CsI(T1)、CsI(Na)、和LiI(Eu)。
NaI(Tl)是使用最广泛的一种闪烁体,它的光产额高,对γ-射线的响应在很大的能量范围内线性,因此它被当做通常γ-谱分析用的标准材料。
作为谱分析探测器的一项重要指标是能量分辨率。衡量这一指标的是探测器测出的能量分布中峰值一半处的全宽度(full width at half maximum,FWHM)。如图10.2-6所示,用NaI(T1)测得的某核素γ射线光电峰位置在能量坐标E0处,峰值为H,H/2高度处峰的宽度为ΔE=E2-E1,则它对该核素能量为E的γ光电峰的FWHM为ΔE,能量分辨率为ΔE/E0。一般的NaI(T1)对137Cs-662keVγ射线的分辨率为10%左右。

图10.2-6 表示能量分辨率的半高峰全宽度(FWHM)
NaI(Tl)的缺点是易潮解,必须密封防潮,性脆易损,不能受机械和热的冲击。它的闪烁衰减时间为230ns,对于快的时间分析和高计数率均带来不便,而且随环境温度升高闪光产额和闪烁衰减时间均会改变,从而分辨率降低。NaI(Tl)有延迟荧光,但光电倍增管的时间常数较快,可将它分辨开来,并用幅度甄别,但对高计数率则是有害的。
CsI是另一种广泛应用的闪烁晶体,它的激活剂有Tl和Na两种,这两种闪烁体的闪烁特性差别很大,CsI单位体积的γ吸收效率较NaI(Tl)稍高,对于尺寸和重量有限制的情况(例如空间实验),这是重要的优点。CsI比NaI不易脆袭,便于加工。CsI(Tl)的最大特点是对于不同的激活粒子有不同的衰减时间,因此可用脉冲形状来区别不同辐射,CsI(Na)的荧光发射谱与NaI(Tl)相似,且光产额比NaI(Tl)高,但缺点是衰减时间较慢。另一种是LiI(Eu)由于它由含有丰度加大的Li同位素可与中子起6Li(n,α)反应,所以主要用于测量中子。
(3)其它无机晶体
BaF2和CsF都是不带激活剂的闪烁材料,BGO自70年代末期以来,应用日益广泛,其主要优点是密度大(7.3g/cm3),铋的原子序数高(=83),因此在现有闪烁体中它单位体积对γ射线的光电吸收几率最高,它具有易于操作和使用方便的机械和化学性能,但缺点是光产额只约为NaI(Tl)的10-20%。它的分辨率比NaI(Tl)差,但是它没有像NaI(Tl)或其它闪烁体中引起余辉的成分,因此在动态扫描,例如CT扫描中它的闪烁能随入射光很快变化。BGO的光输出量及衰减时间随温度上升而变坏。
BaF2是高原子序材料中闪烁衰减时间小于lns的唯一闪烁物质,尽管它的光产额比NaI(Tl)低很多,但是在对探测效率和反应时间都有要求时,它仍不失为一种很好的闪烁材料。但是用于时间测量时必须配以对近紫外区灵敏的光电倍增管,因为BaF2的闪烁光有两个成份,快的近紫外区,慢的波长较长。CsF2的光产额只有NaI(Tl)的约10%,但是衰减时间只有4.4μs,因此也是用于时间分辨测量。
ZnS(Ag)和CaF(Eu)是含有激活剂的无机闪烁体。前者是最早使用的闪烁体,它具有很高的荧光效率,但是只能以多晶的粉末获得,厚度大于25mg/cm时它的多晶层对本身发射的荧光便不透明,因此只能用于α-粒子和其它重离子的测量。后者由于不潮解,化学上不活泼且抗破裂,主要用于恶劣环境中的辐射测量。
3.半导体探测器
闪烁探测器的局限性是能量分辨率差,因为能量转换需经几个步骤,产生一个信息载体需大于100eV的能量,典型射线产生的信息载体数小于几千。半导体探测器提供了改进分辨率的途径,对于给定的入射辐射,它可产生更多的信息载体。它的主要信息载体为电子空穴对,相当于气体中产生的离子对。除此而外,它的体积小,反应时间快,有效厚度大。缺点是体积有限,易受辐射损伤。半导体探测器有时也称为固体探测器,或二极管探测器。作为探测器用的半导体应满足以下几个条件:
①产生电子空穴对的能量尽量小;
②材料中自由载流子应尽量少,以减少噪声;
③载流子俘获中心的数目尽量少,尽量减少对载流子的迁移的影响;
④载流子在材料中的迁移率要高,收集时间要短,以提高探器的时间分辨;
⑤应有高的原子序数Z和较大的密度,以提高辐射与物质的作用几率,从而提高探测效率。
直至现在,能用来作为探测器的半导体材料只有锗和硅的晶体。由于在半导体探测器中产生一个电子空穴对所需要的能量约3.5eV,要比气体中产生一个离子对所需能量低得多,它又不像闪烁探测器那样需要多次转换,因此它的能量分辨率要比其它任何探测器好得多。同样由于产生一有效信号所需的能量较低,它与前级放大器联接后能有较好的信噪比。
(1)半导体探测器的工作原理
半导体探测器实际上由n型和p型半导体单晶紧密联接制成的二极管构成。n型单晶锗和p型单晶锗对接后,n型晶体内束缚较松的电子能较易地穿过p-n结界面与p型晶体的空穴结合,在p-n结周围形成既没有多余电子又没有多余空穴的称做耗尽区的高电阻区。若n型面和p型面分别加上正负电压,即反向电压,则耗尽区增厚,其厚度d与电阻率ρ及所加的偏压的关系为:

图10.2-8为加偏压后p-n结各区的电位分布。加反向偏压的半导体探测器就象一个平板电容,它的电容C取决于探测器灵敏体积的参数:

图10.2-8 在p-n结上加反向偏置后,各区的电位分布
C=KA/(4πW)
式中 A为p-n结的面积;W是耗尽区的宽度;K是半导体材料的介电常数。对于同样的辐射,电容愈小,输出的信号愈大,信噪比也愈高。
目前使用最为广泛的半导体探测器是锂漂移锗(Ge(Li))探测器,它是把锂单晶扩散到锗单晶上制成,现在已能做到灵敏体积在250cm以上。它要在液氮温度下使用保存。由于半导体材料工业的发展,现在已能制出杂质浓度低于10-13的超高纯锗(HPGe)本征锗探测器,其性能可和Ge(Li)相比,并可在室温下保存。Ge(Li)和HPGe探测器结构类型如图10.2-7所示。图10.2-9则示出了一个单开端的Ge(Li)探测器结构。

图10.2-7 Ge(Li)和高纯锗(HPGe)探测器结构示意图

图10.2-9 单开端锂漂移锗探测器
(2)半导体探测器的能量分辨率和探测效率
能量分辨率高是Ge(Li)探测器的主要特点,它对60Co的1.33MeV的光电峰的FW HM可达1.7KeV。由于探测器能量分辨率ΔE/E与一个入射粒子在探测器介质中产生的信息载体(如气体中离子对,半导体中的电子空穴对)数目N的开方成反比,如前所述,对Ge(Li)来说,这个数目比气体和NaI(Tl)都要高,因此它的ΔE/E可比电离室好几倍,比NaI(Tl)谱仪好10-20倍。
Ge(Li)探测器的探测效率低于NaI(T1),它的相对探测效率是与Φ7.6×7.6cm柱型NaI(Tl)探测器对60Co的1.33MeVγ-射线的探测效率相比来确定的,并规定了以下具体测量方法:
①将活度已知的60Co点源放在离Ge(Li)晶体表面25cm处的晶轴位置上;
②测量一段时间直至60Co的1.33MeV光电峰取得足够计数;
③用下式计算Ge(Li)的绝对效率
εab=A·exp(0.693td/T1/2)/(I·p·t)
其中A是1.33MeV光电峰下的总计数,t是测量时间,T1/2是60C。的半衰期,I为以Bq为单位的源活度,p是1.33MeVγ-射线的分支比,td则是由源活度的参考时间至测量时间的时间间隔。
④用下式计算出Ge(Li)探测器的相对探测效率
εr=εab×100%/(1.2×10-3)
其中1.2×10-3为Φ7.6×7.6cmNaI(T1)晶体对距表面25cm出该点源的1.33MeVγ-射线的探测效率。
4.量热计
早在1903年Pierre Curie就曾用量热计测量了镭的放射性。用量热计测量活度,只要求在一定的时间间隔t内测量核衰变所释放的全部能量E,然后除以每次衰变释放的平均能量 ,就能得到测量时间t内的衰变数,不需要知道其它参数。量热计测量方法的灵敏度比用其它方法低得多,所以主要用于强活度测量。
,就能得到测量时间t内的衰变数,不需要知道其它参数。量热计测量方法的灵敏度比用其它方法低得多,所以主要用于强活度测量。
量热计也是测量电离辐射吸收剂量的基本方法,各国一般都以石墨量热计作为γ射线吸收剂量基准。作为吸收剂量测量的量热计与活度测量量热计原理相似,不同之处是射线束来自量热计外部,吸收体处于介质的平衡厚度以后。
以活度测量的量热计——热流型量热计为例,它有三个主要部件:恒温体、放置放射源并吸收其辐射功率的吸收体和热电堆。未放辐射源时,整个系统的温度处于T0。有源时,由于吸收了源的辐射功率W′,使吸收体的温度逐渐上升,同时通过空气和热电堆向恒温体散发热量。由于恒温体的热容很大,它的温度始终保持在T0不变。吸收体所吸收的辐射源功率W′是:
 (10.2-22)
(10.2-22)
式中,A是放射源的活度; 是每次衰变所辐射的平均能量;P为吸收体的吸收系数(对α、β粒子,一般P==1,对γ射线P≤1)。
是每次衰变所辐射的平均能量;P为吸收体的吸收系数(对α、β粒子,一般P==1,对γ射线P≤1)。
若吸收体的热容为C,表面积为S,散热系数为a,温度为T,则可通过热方程式和牛顿冷却定律得到
T-T0=W′[1-exp(aSt/C)]/(aS)
理论上,当t→∞时,exp(-aSt/C)→0,T达到平衡状态的温度Tc,即
Tc-T0=W′/(aS)
若在一段时间内温度上升的变化比测量要求的误差小得多,则我们可认为这时已达到平衡,可将测得的T值当做平衡值Tc。实际上我们测量的并不是温差T-T0,而是热电堆上产生的与其成正比的热电势E1,然后用已知功率为W″=I2R的加热线圈来刻度量热计系统,这时得到热电堆上产生的相应热电热E2,从而得到吸收体吸收辐射源的功率:
W′=(E1/E2)×W″=(E1/E2)×I2R
对于α、β放射源,Pa=Pβ=1,可由公式(10.2-22)求出放射源的活度公式:

5.化学剂量计
化学剂量计是用来测量吸收剂量的一个重要手段。在这种方法中,吸收剂量是由测量介质中所产生的化学变化(包括化合键的变化,以及自由基的变化)来确定的。辐射化学产额是用来表征化学变化量与化学体系吸收辐射能量之间关系的物理量,定义为n(X)除以E的商,其中,n(X)是化学体系从辐射得到平均能量E后,产生、破坏或变化的某一指定成分X的平均物质量。即
G(X)=n(X)/E
辐射化学产额的单位是mol·J-1,通常G的数值是以每l00eV能量产生的某一指定离子的平均数目表示。如硫酸亚铁剂量计在60Coγ射线照射下的G(Fe)值为15.5。不同的化学体系有不同的辐射化学产额。表10.2-2是几种化学体系在60Co辐照下的G值。
表10.2-2几种化学剂量计在60C0辐照下的G值

(1)Fricke剂量计
在化学剂量中目前最可靠、研究最多的是硫酸亚铁剂量计,于1927年由H.Fricke首先提出,所以又叫Fricke剂量计。
标准的Fricke剂量计由1mmol/L的FeSO4或FeSO4(NH4)2SO4·6H2O,lmmol/L的NaCl,0.4mol/L的H2SO4以及含有饱和空气的三次蒸馏水组成。在辐射照射下,Fe2+被氧化成Fe3+,其数目与吸收剂量成正比。可以用分光光度计测量辐照前后在波长304nm处的消光差(εr-ε0),然后按下式计算吸收剂量
D=N(εr-ε0)/(ρ·G·l·εm)
式中,N为阿伏伽德罗常数,6.022×1023mol-1;εm为三价铁离子的克分子消光系数;ρ为化学剂量计的溶液密度;l为分光光度计比色池的光程长度(cm);ε0、εr为照射前后溶液的消光系数。G是Fricke剂量计的Fe3+辐射化学产额。
Fricke剂量计的G值是靠与量热法或电离室法相比较而求得。因此在确定G值的计算中,摩尔消光系数ε和吸收剂量刻度值是影响G值不确定度的主要来源。用Fricke剂量计测量吸收剂量的总不确定度约为(1.5-4%)。
其他体系的化学剂量计性能不如Fricke剂量计稳定、可靠和准确度高。但是可弥补Fricke剂量计适用范围(40-400Gy)窄的缺点。例如硫酸铈剂量计的测量范围是102-2×105Gy,经Fricke剂量计的刻度后,可用于大剂量测量。
(2)丙氨酸/ESR剂量计
根据Pauli不相容原理,一个电子轨道最多只能存在两个自旋相反的电子。电子自旋运动产生的自旋磁矩,对于处在结合形式的每对价电子的净磁矩是零。在辐射作用下,这种对偶电子被打破而产生未偶电子,即被辐照物质产生了自旋磁矩的变化,用电子自旋共振(ESR)谱仪可测出其变化大小。
丙氨酸,特别是L-丙氨酸是现代用得较广、计量学性能最好的一种剂量计材料。它的分子式是

在射线辐照下,丙氨酸分子的C-H键断裂,失去氨基,形成CH3-CH-COOH一个未偶电子,称为自由基。自由基的数目和所吸收的剂量成比例,可借助ERS谱仪测出自由基数目,从而得到吸收剂量,所以丙氨酸/ERS剂量计的实质是自由基型剂量计。
在外磁场作用下,顺磁物质中未偶电子的自旋磁矩有两种不同的取向,形成两个能级E1和E2,其能量差ΔE与Lande因子g、Bohr磁子β和磁场强度H的关系是
ΔE=E2-E1=gβH (10.2-23)
对给定物质,β和g都是常数,因此ΔE就正比于磁场的强度,如果在垂直于外磁场的方向加上一个频率为v的高频电场,并使它满足以下条件:
hv=gβH
则处于E1能级上的部分未偶电子吸收能量hv后,可跃迁到E2能级上,这一过程称“共振吸收”,它是顺磁物质中未偶电子在磁场中吸收一定波长的电磁能而产生共振跃迁的物理现象。
ERS波谱仪由微波发生器产生的高频微波hv经波导系统偶合到放置样品的共振腔。共振腔位于电磁铁两级之间的间隙中。腔内样品对磁场作线性扫描。当改变磁场强度H满足式(10.2-23)的共振条件时,出现自旋共振,样品吸收微波功率hv后,使通过共振腔的微波功率发生变化。用微波检测器检测这一变化,并用放大器将其放大,最后用示波器显示或用记录仪记录,得到ERS信号(如图10.2-10)。图中显示了5个高度不等的峰。中间主峰的幅度最大,它的幅度正比于吸收剂量。

图10.2-10 辐照后的丙氨酸的ESR波谱
绝对测量样品中的自由基浓度很困难,因此ERS剂量计通常是用高一级剂量计先进行标定,再用于吸收剂量的测量,用作量值传递剂量计。它的主要优点是:①适用于累集剂量测量;②剂量测量的线性范围宽,能达到1-104Gy;③读数后,样品并不损失信息,可供以后复查使用。
6.活化探测器
某些核素在俘获一个中子以后,形成原子序数与其相同,但原子质量数大1的同位素,这种新生成的同位素常常是不稳定的,而是处于激发状态,并且通过放射性衰变转变成另一种元素。通过对它在中子场中照射后形成的放射性进行测量,可以测出中子场的中子注量率。因此这种核素可用来测量中子,利用这一原理的中子探测器称为活化探测器。
如果作为探测器的核素在注量率为φ的中子场中照射,核素的平均俘获截面为 ,探测器的总核数为N,且厚度足够薄,生在核素的衰减常数为λ,那么在照射t时间以后探测器的放射性活度为:
,探测器的总核数为N,且厚度足够薄,生在核素的衰减常数为λ,那么在照射t时间以后探测器的放射性活度为:

如果λ值很大,即半衰期很短,则照射足够长时间以后t>10λ-1),即可达饱和活度。若λ值很小,即半衰期很长,当t比λ小得多时,可近似地认为:

选用活化探测片时应考虑以下几点:①对中子的俘获截面比较大;②生成的放射性易于测量;③化学物理性能较稳定,易于保存和加工;④生成的放射性核素半衰期要合适。可以用为活化探测器的核素有23Na,30Si,51V,56Mn,58Fe,63Cu,115In,197Au等,其中197Au,115In,55Mn,23Na等都是经常用的探测器。
金具有许多独特的优点:①具有单一的稳定的同位素197Au;②热中子俘获截面大,且知道很很清楚,在低能区很大的范围内截面遵循1/v规律;③199Au的衰变较简单,便于绝对测量,且半衰期适中;④化学物理性能良好,因此它是测量热中子注量率的最好的活化探测器。锰也有符合做探测器的特点,它的化合物Mn2SO4可溶于作为慢化剂的水中,因此它可作为水慢化剂中的中子指示剂,近代中子源强度的绝对测量几乎无一例外地都用以Mn2SO4水溶液为基础的锰溶法。
【参考文献】:
[1]ICRU,Radiation quantities and units,ICRU,Report 33(1980).
[2]ICRU,Neutron dosimetry for biology and medicine.ICRU,Report 26(1977)
[3]ICRU. Determination of dose equivalents resulting from external radiation sources, Report 39 (1985).
[4]ICRU,Determination of dose equivalents resulting from external radiation sources,Part 2,Report 43(1988).
[5]Quantities and units in radiation protection dosimetry. ICRU,Report 51(1993).
[6]ICRP.1990 Recommendations of the ICRP Publication 60(1991).
[7]ISO—8529, Neutron reference radiation for calibrating neutron measuring devices used for radiation protection purpose and for determining their response as a function of neutron energy.
[8]IEC—1005,Portable neutron ambient dose equivalent ratemeter for use in radiation protection (1990).
[9]IAEA,Guideline on calibration of neutron measuring devices, IAEA Technical Reports Series No. 285(1988).
[10]卢希庭,原子核物理,原子能出版社,1981。
[11]梅镇岳,原子核物理学,科学出版社,1961。
[12]Mann,W. B. ,A handbook of radioactivity measurements procedures,NCRP Report 58(1985).
[13]Johnson C. H. ,Fast Neutron Physics,Part I,Interscience Publisher (1960).
[14]Knoll,G. F. , Radiation Detection and Veasurement, 2nd Ed.,John Wiley & Sons(1989).
[15]Attix,F.H. , Tochlin, E. , Radiation Dosimetry. Vol.1-4,Academic Press ,New York(1968).