烟草化学成分分析
出处:按学科分类—工业技术 广东经济出版社;中国轻工业出版社《烟草工业手册》第788页(101789字)
(一)试样的制备和含水率的测定
1.原理
将烟草或烟草制品经过低温烘干,研磨成一定粒度的烟末。在一定条件下烘干试料,由烘干前后的质量差求出试样中的含水率。
2.仪器
(1)鼓风干燥箱,控温精度±1℃,温度均匀度±1℃。
(2)粉碎机。
(3)筛网,孔径0.45mm(40目)。
(4)硅胶干燥器。
(5)称量皿,磨口具盖,直径40~65mm,深20~45mm。
(6)广口瓶,磨口具塞,容积250mL。
3.试样的制备
若为烟叶,从实验室样品各部分随机抽取一部分烟叶,用软毛刷将烟叶上的细土和沙粒刷去,抽去主脉,将烟叶剪成片或切成丝;若为卷烟,应将卷烟纸和滤材从烟丝中剔除干净。
将烟叶(或烟丝)放入烘箱中,在不高于40℃的温度下烘干,直至可用手指捻碎。
从烘箱中取出烘好的烟叶(或烟丝),马上研磨,持续研磨时间不应超过2min。然后过筛网,将未过筛的细脉重新研磨过筛。
将通过筛网的烟未立即装入洁净干燥的广口瓶中密闭起来。充分摇动,混匀。
4.含水率测定
将编有号码的洁净具盖称量皿打开盖子,一同放入100℃±1℃温度下烘干2h。放入硅胶干燥器内冷却至室温(约30miin),立即称取质量m0,精确至0.001g。向称量皿中加入2~3g试料,称取质量m1,精确至0.001g。将称量皿打开盖子,一同放入烘箱中,每275cm2放置一个称量皿,且只使用干燥箱中央的一层搁板,在100℃±1℃温度下烘干2h。加盖取出称量皿,放入硅胶干燥器内冷却至室温(约30min),称取质量m2,精确至0.001g。
5.计算
按下式计算含水率w:

式中 w——试样的含水率,%(质量分数);
m0——称量皿质量,g;
m1——烘干前称量皿与试料的总质量,g;
m2——烘干后称量皿与试料的总质量,g。
每个试样应平行测定两次,以两次平行测定的平均值作为测定结果,精确至0.01%。两次平行测定结果绝对值之差不应大于0.10%。
(二)烟草中糖的测定
1.水溶性糖的测定——芒森沃克法
(1)原理:溶剂萃取出水溶性糖,经纯化后(水溶性总糖测定时应水解)与费林溶液在一定条件下反应,产生氧化亚铜沉淀。用三价铁离子溶解氧化亚铜沉淀,产生二价铁离子。
用高锰酸钾标准溶液滴定二价铁离子,求出氧化亚铜沉淀中铜的量,查汉蒙表8-4-2得相应的葡萄糖量,计算得出试样的还原糖(或水溶性总糖)含量。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①草酸钾(K2C2O4·H2O)。
②费林溶液。
a.硫酸铜溶液:溶解硫酸铜(CuSO4·5H2O)34.639g于水中,稀释至500mL;
b.酒石酸钾钠溶液:溶解酒石酸钾钠(KNaC4H4O6·4H2O)173.09g及氢氧化钠50g于水中,稀释至500mL。
③3mol/L盐酸溶液。
④醋酸铅溶液:中性醋酸铅[Pb(CH3COO)2·3H2O]溶于水制成饱和溶液。
⑤10%氢氧化钠溶液(质量分数)。
⑥硫酸铁溶液:溶解铁铵矾[FeNH4(SO4)2·12H2O]125g或硫酸铁55g于水中,稀释至1000mL。
⑦2mol/L硫酸溶液。
⑧乙醇溶液,80%(体积分数):乙醇加水,用酒精计调整至乙醇浓度为80%(体积分数)。借助pH试纸,用10%(质量分数)氢氧化钠溶液中和乙醇溶液的游离酸至中性。
⑨草酸钾溶液,20%(质量分数)。
⑩高锰酸钾标准滴定溶液。
⑾邻菲罗啉指示剂:溶解邻菲罗啉0.7425g于25mL0.025mol/L硫酸亚铁溶液中。
⑿0.1%甲基红指示剂。
(3)仪器:
①水浴锅。
②抽滤装置。
③25mL单刻度移液管。
④250mL容量瓶。
⑤250mL三角瓶。
⑥400mL烧杯。
⑦漏斗,直径约为90mm。
⑧500mL圆底烧瓶。
⑨25mL棕色滴定管。
⑩G4型烧结玻璃坩埚。
⑾快速定性滤纸。
(4)方法:
①高锰酸钾标准滴定溶液的制备:
a.配制:称取约3.3g高锰酸钾,溶于105mL水中,缓缓煮沸15min,冷却后置于暗处保存二周,以G4号烧结玻璃坩埚过滤于干燥的棕色瓶中。
注:过滤高锰酸钾溶液所使用的G4号烧结玻璃坩埚预先应以同样的高锰酸钾溶液缓缓煮沸5min,收集瓶也要用此高锰酸钾溶液洗涤2~3次。
b.标定:称取0.2g于105~110℃烘至恒量的基准草酸钠,称准至0.0001g,溶于100mL硫酸溶液(8份硫酸+92份水)中,用配制好的高锰酸钾溶液[c(KMnO4)=0.1mol/L]滴定,近终点时加热至65℃,继续滴定至溶液呈粉红色保持30s。同时做空白实验。
c.计算:按下式计算高锰酸钾标准溶液的滴定度:

式中 T——高锰酸钾标准溶液的滴定度,mgCu/mL;
m——草酸钠的质量,g;
V1——滴定消耗高锰酸钾标准溶液的体积,mL;
V2——空白实验消耗的高锰酸钾标准溶液的体积,mL。
127.1=2×铜的相对原子质量
134.0=草酸钠的相对分子质量
浓度值精确至0.0001mgCu/mL。
平行实验不得少于8次,两人各做4平行,每人4平行测定结果的极差与平均值之比不得大于0.2%;两人测定结果平均值之差与总平均值之比不得大于0.2%。结果取平均值。
②测定:称取烟草试样适量(烤烟约2~3g,晾晒烟约4~6g)于三角瓶中,准确至0.001g,加入100mL乙醇,浸泡过夜。次日,将上层清液过滤于圆烧瓶中,再取100mL乙醇倒入三角瓶中,在沸腾的水浴锅上回流抽提30min。将抽提液过滤于圆底烧瓶中,用乙醇充分洗涤三角瓶及残渣,合并抽提液与洗涤液,加入瓷环2个,在沸腾的水浴锅上回收乙醇,直至没有乙醇味。取下圆底烧瓶,加入100mL80℃水,振荡溶解,必要时用玻璃棒搅碎不溶块,冷却至室温。将溶液转入容量瓶中,用水充分洗涤圆底烧瓶内壁,将洗涤液转入容量瓶中。沿容量瓶壁小心加入醋酸铅溶液15mL,用水定容至刻度,盖上瓶盖,充分振荡,静置15min。然后将溶液过滤于内有2g草酸钾的三角瓶中,待滤液约200mL时,充分摇动三角瓶使草酸钾溶解,静置,用一滴草酸钾溶液检查上层清液应无沉淀产生。将溶液过滤于三角瓶中,此即为制备好的糖液。
用单刻度移液管移取制备好的糖液25mL于烧杯中,加入盐酸溶液3mL,盖上表面皿,放在沸腾的水浴锅上水解,并准确控制水解时间15min,然后将溶液迅速冷却至室温,加甲基红指示剂2滴,用氢氧化钠溶液调节溶液至中性,氢氧化钠溶液不可过量。即刻移取费林溶液a和b各25mL于烧杯中,加水17.5mL。盖上表面皿,将烧杯加热,使之在4min内沸腾,并准确控制溶液保持沸腾2min,立即将氧化亚铜沉淀抽滤于烧结玻璃坩埚内,以60~80℃的水充分洗涤烧杯和烧结玻璃坩埚,将烧结玻璃坩埚放入原烧杯内。
用单刻度移液管移取制备好的糖液和水25mL于烧杯中,然后移取费林溶液a和b各25mL于烧杯中,进行上述操作。
向装有烧结玻璃坩埚的烧杯中加入硫酸铁溶液50mL,搅动溶液使氧化亚铜沉淀完全溶解,然后加入硫酸溶液20mL,立即用高锰酸钾标准滴定溶液滴定,接近终点时,加入邻菲罗啉指示剂1滴,滴定至溶液由棕黄色变成绿色即为终点。同时做空白实验,并加以校正。空白实验有效期为2个月。
(5)计算:
①铜量的计算:
按下式计算生成的铜量:
m=T(V2-V1)
式中 m——生成的铜量,mg;
T——高锰酸钾标准滴定溶液的滴定度,mgCu/mL;
V1——空白实验耗用的高锰酸钾标准滴定溶液的体积,mL;
V2——滴定耗用的高锰酸钾标准滴定溶液的体积,mL。
②水溶性糖含量的计算:
按下式计算烟草中水溶性总糖(或还原糖)的质量分数:

式中 w糖——水溶性总糖的质量分数,%;
N——由表8-4-2查得的Zmg铜相应的葡萄糖量,mg;
m——试样质量,g;
w——试样的含水率,%。
以两次平行测定的平均值作为测定结果。
表8-4-2 汉蒙表 单位:mg



若测得的水溶性糖含量大于或等于10.0%,结果精确至0.1%;若小于10.0%,结果精确至0.01%。
两次平行测定结果绝对值之差不应大于0.30%。
2.直接还原糖的测定
(1)原理:在试样中加入80%乙醇溶液,萃取糖,用醋酸铅除去萃取液中的蛋白质后测定还原能力,用葡萄糖直接还原糖。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①80%乙醇溶液。
②索莫季试剂:将30g酒石酸钾钠(KNaC4H4O6·4H2O)和30g无水碳酸钠溶解在200mL热水中,加入40mL1mol/L的氢氧化钠溶液,再加用30mL水溶解的8g硫酸铜(CuSO4·5H2O),加热15min赶出溶液中的空气。另取180g无水硫酸钠溶解在约500mL热水中,加热排出溶液中的空气。冷却后将上述两溶液移入1L的容量瓶中。另用约10mL的水溶解8g碘化钾,并加适量的碘酸钾,加入上述溶液,冷却后加水到1L。
③中性醋酸铅溶液:将36g中性醋酸铅[Pb(CH3COO)2·3H2O]溶解在95mL水中。
④无水草酸钠。
⑤1mol/L氢氧化钠溶液。
⑥1mol/L硫酸溶液。
⑦0.05mol/L硫代硫酸钠溶液:将25g硫代硫酸钠溶解于1L水中,加热并使其沸腾5min,趁热装入褐色瓶中,于低温避光处保存。该溶液的系数按下述方法求出:
精确称取经130℃干燥1h的特级碘酸钾0.15g,放入200mL的三角瓶中,加水100mL,在加2g碘化钾和1mol/L硫酸溶液15mL,用硫代硫酸钠溶液滴定游离碘,当黄色基本消失时,加入淀粉指示剂。紫色消失时为滴定终点。系数用下式求出:

⑧0.0025mol/L硫代硫酸钠溶液:由于该浓度的溶液不稳定,应先配制0.05mol/L的溶液,准确标定后存放起来,使用前再稀释到0.0025mol/L。
⑨淀粉指示剂:将1g可溶性淀粉放入20mL冷水中搅成糊状,再倒入140mL沸水中,搅匀煮沸5min,加20g氯化钠,冷却后用水定容到200mL。
(3)方法:
①准确称取1.5g试样(晾晒烟及白肋烟为1g),放置于200mL圆底烧瓶或三角烧瓶中,加80%乙醇溶液约100mL,装上回流冷凝装置,在水浴上加热萃取1h。
②冷却后取上层清液,残渣再加100mL乙醇溶液,用上述的方法萃取,再用布氏漏斗过滤,并用80%乙醇溶液清洗残渣数次,最后将上部清液、洗液和滤液合并到一起。
③加热萃取液(最好用减压浓缩),直到没有乙醇味为止,再加适量温水,过滤到100mL的容量瓶中。在滤液中加入中性醋酸铅溶液,直到不出现沉淀为止(约3mL)。充分摇混后加水定容为100mL。
④用滤纸过滤,在滤液中加无水草酸钠,直到不出现沉淀为止。充分搅拌后放置1.5~2h,再用干燥滤纸过滤。
⑤吸取10mL滤液(晾晒烟及白肋烟为25mL)放入100mL容量瓶中,用B.T.B试纸测定pH,中和后加水定容为100mL。
⑥准确吸取上述糖液5mL于25mm×200mm的试管中,加索莫季试剂5mL。
⑦另取一同样试管,分别加入水和试剂各5mL,作为空白对照。
⑧试管口加玻璃球盖,将数支试管放入金属框中,然后在沸腾水浴上加热15miin。
⑨加热完毕后立即用自来水冷却,再加入1mol/L硫酸溶液2mL。硫酸要迅速加入,同时充分摇混使溶液一下成为酸性。
⑩用0.0025mol/L硫代硫酸钠溶液滴定游离碘,当黄色基本消失时,加入淀粉指示剂。紫色消失时为滴定终点。
(4)计算:
按下式计算烤烟的直接还原糖w1,以%表示:

按下式计算晒烟或白肋烟的直接还原糖:

式中 A——5mL试样溶液中的葡萄糖量,g;
m——试样的质量,g。
A值按下述方法求出:用纯的葡萄糖配制成每5mL分别含糖0.5mg、1.0mg、1.5mg、2.0mg、2.5mg的糖液,滴定该糖液即可求出葡萄糖与硫代硫酸钠溶液滴定值的关系式。
3.葡萄糖、果糖、肌醇、蔗糖的测定
(1)原理:单糖、双糖类等可溶性糖,用80%乙醇溶液萃取后,经甲硅烷基化后用气相色谱仪分析。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①80%乙醇溶液。
②甲硅烷基化试剂TMS-PZ。
③内标物山梨糖醇液:准确称取约400mg山梨糖醇,放入100mL容量瓶中,加水定容为100mL。
④糖类校正曲线用溶液:
a.准确称取240mg无水葡萄糖、160mg果糖、50mg肌醇、240mg蔗糖放入100mL容量瓶中,加水定容为100mL。
b.准确吸取50mLa溶液放入100mL容量瓶中,加水定容为100mL。
c.准确吸取10mLa溶液放入100mL容量瓶中,加水定容为100mL。
(3)方法:
①准确称取1g试样放置于300mL茄型烧瓶或三角烧瓶中,约加150mL80%乙醇,装上回流冷凝器在沸水浴上加热1h。冷却后,加山梨糖醇水溶液(烤烟5mL,其他类型烟1mL)用同山式漏斗(No.5A滤纸)过滤。
②取一定量(烤烟50μL,其他类型烟100μL)萃取液放入2mL的试管中,在干燥器中减压干固后,用微量注射器加40μLTMS-PZ,放置约15min后用气相色谱仪分析可溶性糖类。
③气相色谱仪的条件:
气相色谱仪,配有氢火焰离子检测器及数据处理装置。
色谱柱:OV 101,0.25mmi.d.×30m玻璃毛细管;
温度:150~280℃(3℃/min);
载气:He,0.9mL/min;
试样注入量:1μL,感度:10×8。
④测定色谱图中葡萄糖、果糖、肌醇、蔗糖的峰高。
⑤分别吸取校正曲线用溶液各10mL放入三角烧瓶中,再加1mL山梨糖醇内标液,混合后各取20μL分别放入2mL的试管中,在干燥器中减压干固后,用微量注射器分别加入30μLTMS-PZ,放置约15min后用气相色谱仪分析。根据峰高比和质量制作校正曲线。
(4)计算:根据校正曲线计算试样中各类糖的含量。
4.总糖的测定(自动分析仪法)
(1)原理:将黄色赤血盐在碱性环境中用还原糖还原成无色的黄血盐的退色比色法,用自动分析仪进行糖的定量。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①30%Brij35[聚氧化乙烯(23)月桂醚]溶液。
②0.25%硫酸锌和0.25%硫酸铝钾[KAl(SO)4·12H2O]的混合溶液:将5g硫酸锌(ZnSO4·7H2O)和5g特级硫酸铝钾溶解于水中,定容为2L。再加入1mLBrij35。
③0.033mo1/L氢氧化钠溶液:将2.7g氢氧化钠溶解在煮沸后除去二氧化碳的软化水中,定容为2L。再加入1mL30%Brij35。
④0.3mol/L盐酸溶液:将52.4mL浓盐酸加到软化水中,定容为2L。再加入1mLBrij35。
⑤1mol/L氢氧化钠溶液:将40g氢氧化钠溶解在煮沸后除去二氧化碳的软化水中,定容为1L。再加入0.5mL30%Brij35。
⑥0.046%碱性铁氰化钾溶液:将0.92g特级铁氰化钾[赤血盐K3Fe(CN)6]溶解于水中后,加入16g氢氧化钠,溶解后用水定容为2L。再加入1mL30%Brij35。
⑦1%氰化钾溶液:将20g氰化钾溶解于软化水中,定容为2L。再加入1mL30%Brij35。
⑧0.25%苯甲酸溶液:将5g苯甲酸加温溶解于软化水中,定容为2L。
⑨葡萄糖标准原液:将葡萄糖在100℃干燥1h后,精确称取10.000g,溶解在0.25%苯甲酸溶液中,准确定容为1L。
⑩葡萄糖标准溶液:分别吸取原液5mL、10mL、15mL、20mL、25mL、30mL、35mL,用0.25%苯甲酸溶液准确定容为100mL。该标准溶液在100mL中含葡萄糖分别为50mg、100mg、150mg、200mg、250mg、300mg、350mg。
(3)仪器:自动分析仪。该装置由以下各部分组成:
①取样器:自动取样装置,在取样器平板上放有40个取样杯。将其速度调节为每小时输送10个样杯。
②将具有一定内径的输液管安装在平板上,滚轮一面压着输液管一面移动,使溶液以一定的流速输送到下面的装置内。
③混合蛇型管:小型玻璃蛇型管,用于混合2种以上的溶液。
④连续过滤装置。
⑤加热槽:用两根长12m的玻璃蛇型管插入能够调节温度的液槽中,用于加热反应。
⑥透析槽:利用透析除去有害物质。
⑦比色计、记录仪。
(4)方法:
①准确称取约1g试样于具塞三角瓶中,加入100mL软化水,摇振20min,然后过滤。
②将自动分析仪的各试剂输管装入软化水中,关闭取样器软化水阀门,打开泵、比色计的开关,冷却器内通入水。
③分别将配制好放置30min以后的0.25%硫酸锌和0.25%硫酸铝钾的混合溶液、0.033mol/L氢氧化钠溶液、0.3mol/L盐酸溶液、0.046%碱性铁氰化钾溶液、1mol/L氢氧化钠溶液、1%氰化钾溶液用泵吸入。
④在取样器上同时准备好葡萄糖标准溶液和方法①的滤液。
⑤打开过滤装置、取样器、记录仪的开关。
⑥取样结束后,关掉取样器。
⑦10miin后将0.25%硫酸锌和0.25%硫酸铝钾的混合溶液、0.033mol/L氢氧化钠溶液的输液管内换成软化水,15min后在0.3mol/L盐酸溶液输液管内换成软化水。
⑧在装有0.033mol/L氢氧化钠溶液的输液管用0.3mol/L盐酸溶液冲1miin,再放入软化水。该酸性溶液不能进入泵中,应在过滤液开始取样之前拿掉。然后倒掉管中的0.3mol/L盐酸,这时过滤液管用软化水冲洗。
⑨关掉过滤装置。
⑩根据记录,拔掉最后一个试样前两个盛有0.046%碱性铁氰化钾溶液的输液管,放入软化水中。最后一个试样输液管仍按原状不动。
⑾记录纸上记录结束以后,关闭记录仪、比色计。15min后关掉泵以及取样器的软化水和冷却器用水。
⑿根据记录仪上标准溶液的吸光度制作标准曲线,由此求出100mL试样溶液中含的毫克数。
(5)计算
按下式计算总糖的含量w总糖,以%表示:

式中 A——100mL试样溶液中的含糖量,mg;
m——试样的质量,g。
(三)烟草中总氮的测定-克达尔法
1.原理
有机含氮物质中的氮,在浓硫酸及催化剂的作用下,经过强热消化分解,氮被分解为氨,与溶液中过量的硫酸结合成硫酸铵,保留于溶液中。以氨基酸为例,反应式如下:

2NH3+H2SO4=(NH4)2SO4
向消化后的溶液中加入强碱,释放出氨,将氨蒸馏于标准溶液中,用碱标准滴定溶液反滴定,求出试样中的氮量。
2.试剂和材料
所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
(1)氧化汞,红色。
(2)硫酸钾。
(3)锌粒。
(4)95%~98%硫酸。
(5)氢氧化钠-硫代硫酸钠溶液:将500g氢氧化钠和40g硫代硫酸钠(Na2S2)2·5H2O)溶于水,稀释至1000mL。
(6)0.1mol/L硫酸标准滴定溶液。
(7)0.1mol/L氢氧化钠标准滴定溶液。
(8)甲基红指示剂,1%。
3.仪器
(1)500mL克氏烧瓶:500mL。
(2)三角瓶:300mL。
(3)量筒:25mL,100mL,250mL。
(4)单刻度移液管:25mL。
(5)碱式滴定管:25mL。
(6)蒸馏装置。
4.方法
(1)0.1mol/L氢氧化钠标准溶液的配制:
①配制:称取50g氢氧化钠,溶于50mL水中,摇匀,注入密闭容器,放置至溶液清亮。用塑料管虹吸10mL上层清液,注入2000mL煮沸放冷之蒸馏水中,摇匀。
②标定:称取约0.6g于105~110℃烘至恒量的基准邻苯二甲酸氢钾,精确至0.0001g,溶于50mL煮沸放冷之蒸馏水中,加入2滴酚酞指示剂(10g/L),用配制好的氢氧化钠溶液滴定至溶液呈粉红色。同时做空白实验。
③计算:按下式计算氢氧化钠标准滴定溶液浓度:

式中 c(NaOH)——氢氧化钠标准滴定溶液浓度,mol/L;
m——邻苯二甲酸氢钾质量,g;
V1——氢氧化钠标准滴定溶液用量,mL;
V2——空白试验氢氧化钠标准滴定溶液用量,mL;
0.2042——与1.00mL氢氧化钠标准滴定溶液[c(NaOH)=1.000mol/L]相当的以g表示的邻苯二甲酸氢钾的质量。
浓度应精确至0.0001mol/L。
平行实验不得少于8次,两人各做4平行,每人4平行测定结果的极差与平均值之比不得大于0.2%;两人测定结果平均值之差与总平均值之比不得大于0.2%。结果取平均值。
(2)0.1mol/L硫酸标准滴定溶液的配制
①配制:量取6mL95%硫酸,缓缓注入2000mL水中,冷却,摇匀。
②测定:量取25mL配制好的硫酸溶液于50mL煮沸放冷的蒸馏水中,加入2滴酚酞指示剂(10g/L),用氢氧化钠标准滴定溶液滴定至溶液呈粉红色。
③计算:按下式计算硫酸标准滴定溶液浓度:

式中 c(1/2H2SO4)——硫酸标准滴定溶液浓度,mol/L;
V1——氢氧化钠标准滴定溶液用量,mL;
c1——氢氧化钠标准滴定溶液浓度,mol/L;
V2——硫酸标准滴定溶液之用量,mL。
浓度应精确至0.0001mol/L。
平行实验不得少于8次,两人各做4次,每人4平行测定结果的极差与平均值之比不得大于0.2%;两人测定结果平均值之差与总平均值之比不得大于0.2%。结果取平均值。
(3)测定:称取烟草试样1g于克氏烧瓶中,精确至0.001g。加入氧化汞0.7g,硫酸钾10g及硫酸25mL,混合均匀。将克氏烧瓶斜置于定氮架上,缓缓加热,当泡沫停止,溶液澄清后,再迅速煮沸1~1.5h。
冷却,加水250mL,冷却至室温。加锌粒2~3颗,倾斜克氏烧瓶,沿瓶壁小心加入氢氧化钠-硫代硫酸钠溶液100mL,不要摇动,立即连接于蒸馏装置,用内有25mL硫酸标准滴定溶液的三角瓶作接收器(内加3~4滴甲基红指示剂),冷凝管末端应浸入酸液中,小心摇动克氏烧瓶,使内容物充分混合。加热蒸馏直至馏出液达150mL。取下三角瓶,同时用水充分洗涤冷凝管末端。用氢氧化钠标准滴定溶液反滴定过量的酸,溶液由红色变为无色即为终点。同时做空白实验,并加以校正。
5.计算
按下式计算烟草中总氮的质量分数:

式中 wN——烟草中总氮的质量分数,%;
V0——空白实验耗用的硫酸标准滴定溶液的体积,mL;
V1——耗用硫酸标准滴定溶液的体积,mL;
V2——滴定耗用的氢氧化钠标准滴定溶液的体积,mL;
c1——硫酸标准滴定溶液的浓度,mol/L;
c2——氢氧化钠标准滴定溶液的浓度,mol/L;
m——试样质量,g;
w——试样的含水率;
14——氮的摩尔质量,g/mol。
以两次平行测定的平均值作为测定结果,精确至0.01%。两次平行测定结果绝对值之差不应大于0.05%。
(四)烟草中蛋白态氮的测定
1.三氯乙酸法
(1)原理:向试样中加入三氯乙酸溶液使蛋白质沉淀,过滤后将残渣用基耶达法分解,再对氮进行定量。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①5%三氯乙酸溶液。
②硫酸。
③硫酸铜-硫酸钾混合粉末:将硫酸铜(CuSO4·5H2O)和硫酸钾(K2SO4)以1∶9的质量比混合后磨碎。
④30%的氢氧化钠溶液。
⑤0.025mol/L的硫酸溶液:将1.4mL的硫酸(93%~98%)缓慢加入水中,边加边搅拌,最后定容到1L。
⑥0.025mol/L的氢氧化钠溶液:取氢氧化钠100g,加水100mL后放入聚乙烯试剂瓶中,存放一星期使碳酸钠析出。将3.2mL上述溶液预先煮沸。排除CO2后用水稀释到2L。系数用下述方法求出:

式中 F——0.025mol/L氢氧化钠溶液的系数。
⑦甲基红-亚甲蓝混合指示剂:将0.20g甲基红溶解在100mL95%乙醇中,制成第一溶液;再将0.10g亚甲蓝溶解在100mL95%乙醇中,制成第二溶液;最后将一份甲基红溶液和一份亚甲蓝溶液混合即制成甲基红-亚甲蓝混合指示剂。
(3)方法:
①准确称取0.5~1.0g试样,加入100mL5%的三氯乙酸溶液,定时摇动,在室温下放置2h以上(一昼夜以内)。
②将沉淀用滤纸过滤,再用5%的三氯乙酸溶液洗涤数次,用液量约150mL。
③将沉淀物和滤纸在60℃下干燥数小时。
④将沉淀物和滤纸一起放入基耶达法烧瓶,向烧瓶中加入约5g的硫酸铜-硫酸钾混合粉末,再加约10mL的硫酸,充分混合。待泡末消失后,用微火加热,逐渐加大火焰,使其分解约3h。
⑤将试样冷却,移入100mL的容量瓶中,加水到刻度。
⑥用移液管从容量瓶中准确吸取20mL溶液,从蒸馏装置的进样口注入,然后用蒸馏水冲洗进样口残存的试样。再从进样口注入30%的氢氧化钠溶液8mL,并用水冲洗进样口。
⑦准确吸取0.05mol/L的硫酸5mL,注入放置在接受器皿中,并将冷却器出口端浸没在硫酸溶液中,蒸馏约10min即可将氨全部蒸馏出。
⑧蒸出液中的过量硫酸用甲基红-亚甲蓝混合指示剂和0.025mol/L的氢氧化钠溶液滴定。
测定的同时应做空白试验。
(4)计算:
用下式计算蛋白态氮:

式中 w蛋白态氮——蛋白态氮的质量分数,%;
V1——消耗的0.025mol/L的氢氧化钠溶液的体积,mL;
V2——空白试验中所消耗的0.025mol/L的氢氧化钠溶液的体积,mL;
F——0.025mol/L氢氧化钠溶液的系数;
m——试样的质量,g。
2.莫尔法
(1)原理:用热醋酸萃取试样,过滤后将残渣用基耶达法分解,再对氮进行定量。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①5%醋酸溶液。
②硫酸。
③硫酸铜-硫酸钾混合粉末:将硫酸铜(CuSO4·5H2O)和硫酸钾(K2SO4)以1∶9的质量比混合后磨碎。
④30%的氢氧化钠溶液。
⑤0.025mol/L的硫酸溶液:将1.4mL的硫酸(93%~98%)缓慢加入水中,边加边搅拌,最后定容到1L。
⑥0.025mol/L的氢氧化钠溶液:取氢氧化钠100g,加水100mL后放入聚乙烯试剂瓶中,存放一星期使碳酸钠析出。将3.2mL上述溶液预先煮沸。排除CO2后用水稀释到2L。系数用下述方法求出:

式中 F——0.025mol/L氢氧化钠溶液的系数。
⑦甲基红-亚甲蓝混合指示剂:将0.20g甲基红溶解在100mL95%乙醇中,制成第一溶液;再将0.10g亚甲蓝溶解在100mL95%乙醇中,制成第二溶液;最后将一份甲基红溶液和一份亚甲蓝溶液混合即制成甲基红-亚甲蓝混合指示剂。
(3)方法:
①准确称取0.5~1.0g试样,放入200~300mL的烧杯中,再加75mL的5%的醋酸溶液,沸腾后继续煮沸5min。
②将沉淀用滤纸过滤,残渣用5%的醋酸溶液洗涤数次,用液量约150mL。
③将沉淀物和滤纸一起放入基耶达法烧瓶,向烧瓶中加入约5g的硫酸铜-硫酸钾混合粉末,再加约10mL的硫酸,充分混合。待泡末消失后,用微火加热,逐渐加大火焰,使其分解约3h。
④将试样冷却,移入100mL的容量瓶中,加水到刻度。
⑤用移液管从容量瓶中准确吸取20mL溶液,从蒸馏装置的进样口注入,然后用蒸馏水冲洗进样口残存的试样。再从进样口注入30%的氢氧化钠溶液8mL,并用水冲洗进样口。
⑥准确吸取0.05mol/L的硫酸5mL,注入放置在接受器皿中,并将冷却器出口端浸没在硫酸溶液中,蒸馏约10min即可将氨全部蒸馏出。
⑦蒸出液中的过量硫酸用甲基红-亚甲蓝混合指示剂和0.025mol/L的氢氧化钠溶液滴定。
测定的同时应做空白试验。
(4)计算:用下式计算蛋白态氮:

式中 w蛋白态氮——蛋白态氮的质量分数,%;
V1——消耗的0.025mol/L的氢氧化钠溶液的毫升数;
V2——空白试验中所消耗的0.025mol/L的氢氧化钠溶液的毫升数;
F——0.025mol/L氢氧化钠溶液的系数;
m——试样的质量,g。
(五)烟草中硝态氮的测定
1.二甲基酚法
(1)原理:用三氯乙酸除去试样水萃取液中蛋白后,加二甲基酚。试样中的硝酸将其硝化生成色素,蒸馏后进行比色定量。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①3,4-二甲基酚:将2.5g的3,4-二甲基酚溶解在100mL的0.2mol/L的NaOH溶液中。
②10%的三氯乙酸溶液。
③80%的硫酸溶液:将72mL的硫酸和28mL的水混合制成。
④0.2mol/L的氢氧化钠溶液。
⑤硝态氮的标准原液:准确称取7.219g硝酸钾放入1L的容量瓶中,加少量水溶解后,再加水至1L。每1mL该溶液含硝态氮1mg。
⑥硝态氮的标准溶液:取一定量的硝态氮标准原液,从0~6mL之间任选数档,分别放入200mL的容量瓶中,各加25mL的10%三氯乙酸,用水定容到200mL。
(3)仪器:分光光度计或光电比色计。
(4)方法:
①准确称取1g的试样,放入200mL的容量瓶中,加150mL的水,用振荡器振荡30min。再加25mL的10%三氯乙酸,充分振荡3min,用水定容,混合后用干燥滤纸过滤(开始的10mL不要)。
②在250mL的圆底烧瓶中放入10mL滤液(硝态氮含量在0.8%以上时,放入5mL滤液和1.25%的三氯乙酸),再加2mL3,4-二甲基酚溶液和40mL的80%硫酸溶液。
③立即将烧瓶塞上塞子充分混合,在室温下放置30min。然后用100mL水稀释,加沸石进行蒸馏。接通垂直的李比希冷凝管流入预先放入的10mL0.2mol/L的氢氧化钠的容量瓶中,约收集50mL的蒸馏液后,停止蒸馏。
④用15mL0.2mol/L的氢氧化钠溶液和水冲洗冷凝管中的色素,并与蒸馏液混合,用水定容到100mL。
⑤在波长432nm处,用1cm的比色皿测定所得溶液和空白试验溶液的吸光度。如果吸光度在0.1以下,用厚比色皿测定吸光度。
⑥按上述方法进行操作,制作硝态氮标准溶液标准曲线,从试样的吸光度求得硝态氮。
(5)计算:使用10mL滤液时,按下式计算硝态氮的质量分数:

式中 w硝态氮——硝态氮的质量分数,%;
m2——被测液的硝态氮量,mg;
m1——试样的质量,g。
2.离子活度计法
(1)原理:用水萃取试样中的硝酸根离子,再用Al离子交换树脂除去萃取液中存在的有机酸、碳酸等干扰物质。离子计是用硝酸离子电极测定硝酸离子。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①磷酸缓冲溶液(0.1mol/L,KH2PO4):准确称取13.609g的磷酸二氢钾放入1L的容量瓶中,用少量水溶解后加水到刻度。
②Al树脂:将130g的硫酸铝[A12(SO4)3·18H2O]溶解于500mL水,加入50mL的离子交换树脂(Dowex 50X8H型),混合后过滤,用水洗净。过滤在低负压下进行,树脂除去重力水后以湿润状态密闭保存在圆柱形容器内。
③硝态氮标准原液:准确称取0.7219g硝酸钾于1L的容量瓶中,用少量水溶解后加水到刻度。1mL该溶液含硝态氮0.1mg。
④硝态氮标准溶液:依次吸取硝态氮标准原液1mL、2mL、3mL、5mL、10mL、20mL,分别放入100mL容量瓶中,然后各加10mL磷酸缓冲溶液,加水定容到100mL。该溶液含硝态氮分别为1×10-6mg/kg、2×10-6mg/kg、3×10-6mg/kg、5×10-6mg/kg、10×10-6mg/kg、20×10-6mg/kg。
(3)仪器、设备:离子活度计(或带刻度放大装置的pH酸度计)及硝酸离子电极和甘汞标准电极。
(4)方法:
①准确称取0.4g试样放入100mL三角瓶中,加50mL水和1mLAl树脂,加盖振动15min。
②用滤纸过滤,滤液接收在100mL的烧杯中。
③在滤液中插入离子电极,启动磁性搅拌器,测定电动势。
④测定系列浓度的标准溶液的电动势,并以半对数底的对数边作为浓度(1×10-6mg/kg),等间隔边为电动势(mV),制成标准曲线,再用电动势的测定结果求出试样的硝态氮。
(5)计算:
用下式计算硝态氮:

式中 w硝态氮——硝态氮的质量分数,%;
X——从标准曲线求出试样的硝态氮浓度,10-6mg/kg;
m——试样的质量,g。
3.离子活度剂法(硫酸铜溶液萃取法)
(1)原理:用硫酸铜溶液并添加硫酸银萃取试样,以消除萃取硝酸根离子时共存的盐类影响。另外参比电极的内参比液使用硫酸钠,以消除氯化物的影响。然后用离子活度计测定硝态氮。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①0.1mol/L硫酸铜溶液。
②硫酸银。
③硝态氮标准原液:准确称取0.7219g硝酸钾于1L的容量瓶中,用少量水溶解后加水到刻度。1mL该溶液含硝态氮0.1mg。
④硝态氮标准溶液:依次吸取硝态氮标准原液1mL、2mL、3mL、5mL、10mL、20mL,分别放入100mL容量瓶中,然后各加10mL磷酸缓冲溶液,加水定容到100mL。该溶液含硝态氮分别为1×10-6mg/kg、2×10-6mg/kg、3×10-6mg/kg、5×10-6mg/kg、10×10-6mg/kg、20×10-6mg/kg。
(3)仪器、设备:离子计及硝酸离子电极及参比电极。参比电极使用甘汞标准电极。内参比溶液用0.25mol/L的硫酸钠。
(4)方法:
①称取0.1~1.0g试样放入烧杯中,加25mL0.1mol/L的硫酸铜溶液,然后用石蜡封口,搅拌1h。
②加硫酸银100mg,用磁性搅拌器搅拌,在条件稳定情况下,一边搅拌,一边读出电动势。
③测定系列浓度的标准溶液的电动势,并以半对数底的对数边作为浓度(1×10-6mg/kg),等间隔边为电动势(mV),制成标准曲线,再用电动势的测定结果求出试样的硝态氮。
(5)计算:
用下式计算蛋硝态氮:
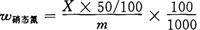
式中 w硝态氮——硝态氮的质量分数,%;
X——从标准曲线求出试样的硝态氮浓度,10-6mg/kg;
m——试样的质量,g。
(六)烟草中氨态氮的测定
1.通气蒸馏法
(1)原理:在烟草的乙醇萃取液中加入硅钨酸,滤掉生物碱后,将滤液进行碱式蒸馏,馏出液中的氨态氮用滴定法或靛酚法定量。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①60%乙醇。
②12%的硅钨酸溶液。
③0.005mol/LH2SO4溶液。
④0.01mol/LNaOH溶液。
⑤苯二甲酸氢钾标准溶液:将标准苯二甲酸氢钾试剂在100~110℃温度下干燥3h,再准确称取20.442g,溶于水后,加水至1L(0.1mol/L溶液)。准确量取10mL该溶液,加水至100mL(0.01mol/L溶液)。
⑥次氯酸溶液:如使用有效氯含量为3%~5%的漂白粉可直接用于试验,如使用有效氯含量为10%的次氯酸标准品时,可加2倍水稀释后使用。
⑦碱性苯酚溶液:将5g苯酚和2g氢氧化钠溶解在100mL水中。
⑧硝普酸溶液:将50mg硝普酸钠溶解在100mL水中;
⑨磷酸缓冲溶液:称取35.82g磷酸氢钠溶解在水中,加水至1L,用氢氧化钠(1mol/L)滴定至pH9.8。
⑩饱和碳酸钾溶液或饱和碳酸钠溶液。
⑾氨态氮标准溶液:准确称取4.7196g硫酸铵,放入1L的容量瓶中,加少量水溶解后,定容至1L。每1mL该溶液含1mg氮。用该溶液制备氮量为2×10-6mg/kg、4×10-6mg/kg、8×10-6mg/kg、10×10-6mg/kg的溶液。
⑿甲基红亚甲蓝混合指示剂:将0.20g甲基红溶解在100mL乙醇(95%)中,制成第一溶液;再将0.10g亚甲蓝溶解在100mL乙醇(95%)中,为第二溶液。最后将一份甲基红和-份亚甲蓝溶液混合制成指示剂。
(3)仪器:使用见图8-4-1所示的通汽蒸馏装置。

图8-4-1 通汽蒸馏装置
A-洗气瓶(0.05mol/LH2SO4溶液) B-试样(试样溶液) C-收集瓶(0.005mol/L硫酸溶液)
a-玻璃球过滤器
(4)方法:
①准确称取1g的试样,加60%的乙醇(约50mL)在室温下进行萃取(搅拌或振荡约30min),然后减压过滤,充分洗净残渣。将残渣再萃取,再过滤。将滤液合并在一起,在40℃以下的温度减压浓缩。
②在浓缩后的溶液中加盐酸使其浓度约为0.3%,然后加12%的硅钨酸溶液直到生物碱完全沉淀为止,放置一夜后过滤。
③在滤液中加饱和碳酸钾溶液或饱和碳酸钠溶液至呈碱性后,用通汽蒸馏装置,并通入不含氨的空气蒸馏,将产生的氨用0.01mol/L氢氧化钠溶液捕集。
④滴定法:捕集液中过量的硫酸,以甲基红亚甲蓝混合指示剂,用0.01mol/L氢氧化钠溶液滴定除去。
⑤靛酚法:将捕集液用50~200mL的容量瓶定容,取1mL经定容的捕集液,依次加入1mL碱性苯酚,7mL磷酸缓冲溶液,0.5mL硝酸溶液,再加0.5mL次氯酸溶液,混合。在室温下放置30min以上,测定650mm的吸光度。用氨态氮的标准溶液做出标准曲线,根据被测物的吸光度求出试样中的氨态氮。
(5)计算:
①采用滴定法时,按下式计算氨态氮的百分含量

式中 w氨态氮——氨态氮的质量分数,%;
V1——试验中用于中和所用的0.01mol/L氢氧化钠溶液的毫升数;
V2——对照试验中用于中和所用的0.01mol/L氢氧化钠溶液的毫升数;
F——0.01mol/LNaOH的系数;
m——试样的质量,g。
②采用靛酚法时,按下式计算氨态氮的质量分数:
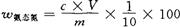
式中 w氨态氮——氨态氮的质量分数,%;
c——被测液氨态氮的浓度,10-6mg/kg;
V——蒸馏液定容的毫升数;
m——试样的质量,g。
2.康怀微量扩散法
(1)原理:将试样萃取后,用离子交换树脂得到的氨组分,在微量扩散分析器中进行氨的扩散和吸收。离子交换树脂的制备方法,有一部分与酰胺态氮的试样制备是相同的,因此请特别注意。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①60%乙醇。
②磷酸缓冲溶液(0.2mol/L,pH7.40):称取27.22g磷酸二氢钾溶解在水中,再用氢氧化钠溶液(约1mol/L)滴定至pH7.40,加水至1L。
③碱溶液:在烧杯中放入110g无水碳酸钾,溶解在100mL的水中,冷却后移入三角烧瓶中,加入沸石,先用强火煮沸,转用微火煮沸约10min。
④0.0025mol/LH2SO4溶液。
⑤1mol/L的KCl溶液。
⑥金和欢胶粘接剂:它是一种用100g金和欢胶粉末,150mL水,50mL丙三醇,50mL碳酸钾饱和溶液的混合物。先将金和欢胶粉末放入大乳钵中,然后徐徐加水,为了促进胶的溶解可以搅拌。然后再加入丙三醇和饱和碳酸钾溶液并搅拌。移入分液漏斗静置一昼夜后,取出下部溶液,放在试剂瓶中贮存。将上部有泡沫的溶液倒掉。
⑦吸收剂:将0.033g的溴甲酚绿和0.066g的甲基红用100mL99%的乙醇溶解,制成混合指示剂。再用200mL99%的乙醇和700mL蒸馏水的混合液中溶解20g的硼酸。再在该溶液中加20mL混合指示剂,充分混合后小心滴入0.05mol/L的氢氧化钠溶液,掌握溶液的颜色恰好到淡粉色那一点(相当于石蕊标准变色表中pH5.4~5.5的颜色,玻璃电极pH3.5~3.6)为止,加蒸馏水到1L。取1mL该吸收剂溶液,加1mL蒸馏水,此时溶液的颜色也必须呈带红的淡褐色。如果呈淡绿色,说明0.05mol/L的氢氧化钠溶液加过量了。因此最好与其他新制备的尚未中和以前的硼酸溶液合在一起再中和。
(3)仪器:
①用异丁烯酸树脂制的康怀微量扩散分析装置(见图8-4-2)。

图8-4-2 康怀微量扩散分析装置
②搅拌棒或搅拌器。
a.搅拌棒:滴定时搅拌的器具,需同样用异丁烯酸树脂制品,其直径为4~5mm的圆棒,形状与规格见图8-4-3所示。

图8-4-3 搅拌棒
b.搅拌器:可代替搅拌棒,滴定时使用起来更为方便。此时要用电磁搅拌器。搅拌器应用四氟乙烯涂料制成,尺寸应能在分析器内室中使用。
c.柱:应使用内径为12mm,并带有旋塞的玻璃柱。
(4)方法:
①制作离子交换树脂柱
a.离子交换树脂的预处理:将约50g的Domex 50-X8(200~400目/25.4mm2)放入500mL的烧杯中,加水400mL,用玻璃棒充分搅拌后静置。树脂沉淀后,将没有沉淀的树脂和上部清液一起倒掉。反复操作3次。
在粒度均一的树脂中加入约3倍量的氢氧化钠溶液(约1mol/L),充分搅拌后静置。约15min后倒掉上部澄清液。然后加水,进行相同处理,以清洗过量的碱(直到上部澄清液基本呈中性为止,需反复数次)。基本达到中性后,加入约3倍量的盐酸溶液(约1mol/L),充分搅拌后,静置约15min后倒掉上部澄清液。然后加水搅拌,再倒掉上部澄清液(反复操作数次,直到上部澄清液基本呈中性为止)。然后再加入上述的氢氧化钠溶液,进行同样处理。倒掉上部澄清液后再加氢氧化钠溶液进行同样操作。倒掉上部澄清液,再反复进行与上述相同的水洗操作,直到溶液呈中性为止(石蕊试纸无反应)。
在所得到的树脂中加300mL磷酸缓冲溶液,搅拌后放置10min,倒掉上部澄清液,反复操作3次。为清除过量缓冲液,需加软化水,进行水洗(3次)。
将经上述处理的离子交换树脂浸泡在软化水中,用带塞的容器存放。如经较长时间才使用,则应放在冰箱中保存。
b.装柱:使用带玻璃滤片的柱子时,柱子内要先装入水,再倒入树脂悬浊液,使树脂部分高度为21mm。使用不带玻璃滤片的柱子时,先加水,然后加入玻璃纤维,再将树脂悬浊液注入。
②准确称取1g的试样,加60%的乙醇(约50mL)在室温下进行萃取(搅拌或振荡约30min),然后减压过滤,充分洗净残渣。将残渣再萃取,再过滤。将滤液合并在一起,在40℃以下的温度减压浓缩。
③将浓缩液用水定容到10mL后过滤。
④取5mL滤液注入已制好的柱子中,滤液如在滤纸上端停止,可用少量软化水洗净玻璃管壁后,用约20mL软化水溶出,用25mL的容量瓶接受溶出液。只接取20~24mL溶出液。该水溶出液的组分,是用于分析酰氨态氮的。
⑤再用1mol/L的氯化钾溶液20mL溶出,溶出液用25mL的容量瓶接取。溶出后加软化水到25mL,此液用于氨的定量。
⑥从溶出液中取出一定量溶液,放入微量扩散分析仪的外室。液量以1~3mL为宜,加水稀释1倍。
⑦在微量扩散分析仪的内室放入1mL吸收剂溶液。然后在分析仪的周沿上涂上金和欢胶粘接剂,再小心往外室加大约3mL的碱悬浊液,加入后马上盖上盖子,再用夹具夹紧。
在水平面上缓缓转动,使外室的溶液完全混合。不要转动太激烈,这样容易使内、外室的溶液混合,也不会溅到盖子上。
⑧将微量扩散分析仪放入25℃的分析室,直到氨完全扩散完为止(24~25h)。
⑨取出微量扩散分析仪,拿掉盖子后,在内室中加1mL蒸馏水。然后用0.0025mol/L的硫酸溶液滴定。滴定时要不断搅动内室的溶液,当溶液的颜色呈甲基红的淡粉色(标准变色表pH为5.0~5.1)时即为终点。
⑩正式滴定前应先进行对照试验,在正式滴定时要不断与对照试验的终点颜色比照。此外,滴定时常用硫酸铵标准溶液(50μg/mL或100μg/mL为宜)及只加试剂不加试样的溶液进行对照试验,对照空白试验至少做3次,并需与试样滤液的定量同时进行。
(5)计算:滴定法可用下式计算氨态氮的质量分数:

式中 w氨态氮——氨态氮的质量分数,%;
V1——对照试验中中和所用的0.0025mol/L硫酸溶液的毫升数;
V2——中和被测液所需的0.0025mol/L硫酸溶液的毫升数;
F——0.0025mol/L硫酸的系数;
V3——微量扩散分析仪外室的试样容量,mL;
m——试样的质量,g。
(七)烟草中酰氨态氮的测定
1.原理
从试样的60%萃取液中经离子交换树脂得到的酰氨组分,再将其加含水率解成氨以后,用通气蒸馏法或微量扩散分析法定量。
2.试剂和材料
所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
(1)3mol/L硫酸溶液。
(2)60%乙醇。
(3)磷酸缓冲溶液(0.1mol/L,pH9.8):称取35.82g磷酸氢钠(Na2HPO4·12H2O)溶解在水中,加水至1L,用氢氧化钠(1mol/L)滴定至pH9.8。
(4)0.0025mol/LH2SO4溶液。
(5)0.01mol/LNaOH溶液。
(6)硝酸溶液:将50mg硝酸钠溶解在100mL水中。
(7)次氯酸溶液:如使用有效氯含量为3%~5%的漂白粉可直接用于试验,如使用有效氯含量为10%的次氯酸标准品时,可加2倍水稀释后使用。
(8)甲基红亚甲基蓝混合指示剂:将0.20g甲基红溶解在100mL乙醇(95%)中,制成第一溶液;再将0.10g亚甲基蓝溶解在100mL乙醇(95%)中,为第二溶液。最后将一份甲基红和一份亚甲基蓝溶液混合制成指示剂。
3.方法
(1)准确称取1g的试样,加60%的乙醇(约50mL)在室温下进行萃取(搅拌或振荡约30min),然后减压过滤,充分洗净残渣。将残渣再萃取,再过滤。将滤液合并在一起,在40℃以下的温度减压浓缩。
(2)将浓缩液用水定容到10mL后过滤。
(3)取5mL滤液注入已制好的柱子中,滤液如在滤纸上端停止,可用少量软化水洗净玻璃管壁后,用约20mL软化水溶出,用25mL的容量瓶接受溶出液。只接取20~24mL溶出液。
(4)从溶出液中取出一定量溶液,移入试管内,再加入3mol/LH2SO4溶液,使最终浓度为0.5mol/L。
(5)盖上盖后,在沸水浴上加热2h。
(6)放冷后取一定量,用氨态氮的通汽蒸馏法或微量扩散分析法进行定量。
4.计算
用定量法的计算公式求出氨态氮,再换算成酰氨态氮。
(八)烟草中氨基酸的测定
1.原理
将试样萃取液中的游离氨基酸用离子交换层析法分离后,用茚满三酮反应定量。
2.试剂和材料
所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
(1)70%乙醇。
(2)缓冲液及茚满三酮液根据分析仪的要求制备。
3.仪器
氨基酸自动分析仪。
4.方法
(1)称取粉末状试样0.5~1.0g,加入70%乙醇溶液约50mL。在室温下搅拌、振动,萃取30min。
(2)减压过滤,并用70%乙醇溶液冲洗,得到滤液和残渣。回收残渣,按上述步骤继续萃取和过滤,得到滤液和残渣。
(3)将2次滤液合并,在40℃以下减压浓缩。
(4)干固后,将其溶解在缓冲液中。在10mL或25mL的容量瓶中用缓冲液定容。
(5)从定容后的溶液中取出所需量,用高速离心(10000~15000r/min)或者用装有微孔过滤片的注射器除去浮游物和不溶物。
(6)以上所得的试样用氨基酸自动分析仪分析。
从分析结果求出各种氨基酸的含量,如有必要,可用氨素含量表示。
(九)烟草中总植物碱的测定
1.紫外分光光度法
(1)原理:烟草试样在强碱性条件下进行水蒸气蒸馏,紫外分光光度法测定定馏出液的吸光度,计算出总植物碱含量,以烟碱表示。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①氢氧化钠,片状。
②氯化钠。
③烟碱,最低纯度98%。
④1mol/LH2SO4。
⑤0.025mol/LH2SO4。
(3)仪器:
①分光光度计:具有230~290nm的波长范围匹配的石英比色皿,光径1cm;
②水蒸气蒸馏装置;
③容量瓶,50mL,250mL;
④单刻度移液管,5mL,10mL;
⑤玻璃漏斗,直径约55mm;
⑥快速定性滤纸。
(4)方法:称取烟草试样1g于蒸馏瓶中,加入20g氯化钠和2g氢氧化钠,立即将蒸馏瓶连接于水蒸气蒸馏装置进行蒸馏,用内含10mL1mol/L硫酸溶液的250mL三角瓶作接收器,冷凝管末端应浸入硫酸溶液中。蒸馏过程中蒸馏瓶内的液体体积应保持恒定,必要时可适当加热。收集220~230mL馏出液,取下容量瓶,同时用水冲洗冷凝管末端。确认容量瓶处于室温,用水定容至刻度。摇匀。若馏出液不澄清,则将其过滤。
移取一定体积(通常为10mL)的馏出液于50mL容量瓶中,用0.025mol/L硫酸溶液定容至刻度。
以0.025mol/LH2SO4溶液为参比,用光谱仪测定溶液在236nm、259nm和282nm处的吸光度,若259nm处的吸光度大于0.7,应取较小体积的馏出液重新稀释测定。
(5)计算:按下式计算烟草中总植物碱的质量分数:

式中 wz——烟草中总植物碱的质量分数,%;
F——稀释倍数;
A236、A259、A282——馏出液在236nm、259nm和282nm处的吸光度的实测值;
m——试样质量,g;
w——试样的含水率,%。
以两次平行测定的平均值作为测定结果,精确至0.01%。两次平行测定结果之差不应大于0.05%。
结果以平行测定的平均值报出,精确至0.01%。
2.添加活性炭法
(1)原理:将活性炭粉末加入烟草试样中,用稀盐酸萃取烟碱,萃取液中的烟碱的定量方法同紫外吸收光谱法,从紫外部吸收来定量。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①活性炭粉末:使用DarcoG-60。
②0.5mol/L盐酸。
(3)仪器:使用波长精度具有2nm以上的分光光度计。
(4)方法:
①准确称取烟草试样0.07~0.2g,用广口漏斗将试样倒入250mL容量瓶中。
②测定烤烟和香料烟时,需往容量瓶内加进试样量2倍的活性炭粉末。
③加入25mL0.5mol/LHCl溶液,用振荡器振摇5min。
④振摇后用水定容,再用滤纸过滤,去掉开始约10mL的滤液,再过滤30mL备用。
⑤用分光光度计测定滤液在236nm、259nm、282nm处的吸光度。
(5)计算
按下式计算烟草中总植物碱质量分数:
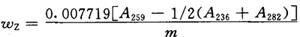
式中 wz——烟草中总植物碱的质量分数,%;
A236、A259、A282——馏出液在236nm、259nm和282nm处的吸光度;
0.007719——250mL萃取液中总碱含量的系数;
m——试样质量,g。
应进行平行测定,平行测定的两个实验数据之间的相对偏差不应大于3%。
结果以平行测定的平均值报出,精确至0.01%。
(十)烟草中总挥发碱的测定
1.原理
烟草试样在pH=8的条件下进行水蒸气蒸馏,试样中的总挥发碱被蒸出,接收于盐酸标准滴定溶液中,用氢氧化钠标准滴定溶液滴定求出总挥发碱的含量,以氨计。
2.试剂和材料
所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
(1)0.1mol/L氢氧化钠标准滴定溶液。
(2)0.1mol/L盐酸标准滴定溶液。
(3)磷酸盐缓冲溶液:将95g氢氧化钾溶于400~500mL煮沸放冷的蒸馏水中,加入氢氧化钡饱和溶液至无沉淀产生为止,滤去沉淀。搅拌下缓缓将350mL磷酸溶液(56份磷酸+300份水)加入到氢氧化钾溶液中,冷却。借助于酸度计,用少量磷酸或氢氧化钾溶液调节溶液pH为8.0。
(4)甲基红加次甲基蓝混合指示剂:0.5%甲基红乙醇溶液与0.25%次甲基蓝乙醇溶液等体积混合。
3.仪器
①水蒸气蒸馏装置,用蒸馏水发生水蒸气。
②酸度计。
③克氏烧瓶,500mL。
④三角瓶,1000mL。
⑤单刻度移液管,15mL。
⑥碱式滴定管,25mL。
4.方法
(1)0.1mol/LNaOH标准溶液的配制:
①配制:称取50g氢氧化钠,溶于50mL水中,摇匀,注入密闭容器,放置至溶液清亮。用塑料管虹吸10mL上层清液,注入2000mL煮沸放冷之蒸馏水中,摇匀。
②标定:称取约0.6g于105~110℃烘至恒量的基准邻苯二甲酸氢钾,精确至0.0001g,溶于50mL煮沸放冷之蒸馏水中,加入2滴酚酞指示剂(10g/L),用配制好的氢氧化钠溶液滴定至溶液呈粉红色。同时做空白实验。
③计算:
按下式计算氢氧化钠标准滴定溶液浓度:

式中 c(NaOH)——氢氧化钠标准滴定溶液浓度,mol/L;
m——邻苯二甲酸氢钾质量,g;
V1——氢氧化钠标准滴定溶液用量,mL;
V2——空白试验氢氧化钠标准滴定溶液用量,mL;
0.2042——与1.00mL氢氧化钠标准滴定溶液[c(NaOH)=1.000mol/L]相
当的以g表示的邻苯二甲酸氢钾的质量。
浓度应精确至0.0001mol/L。
平行实验不得少于8次,两人各做4次,每人4次平行测定结果的极差与平均值之比不得大于0.2%;两人测定结果平均值之差与总平均值之比不得大于0.2%。结果取平均值。
(2)盐酸标准滴定溶液的配制:
①配制:量取9mL浓盐酸,注入1000mL水中,摇匀。
②标定:移取25mL配制好的盐酸溶液于50mL煮沸放冷的蒸馏水中,加2滴酚酞指示剂(10g/L),用0.1mol/L氢氧化钠标准滴定溶液滴定至溶液呈粉红色。
⑧计算:按下式计算盐酸标准滴定溶液:

式中 c(HCl)——盐酸标准滴定溶液浓度,mol/L;
V1——氢氧化钠标准滴定溶液用量,mL;
c1——氢氧化钠标准滴定溶液浓度,mol/L;
V2——盐酸标准滴定溶液用量,mL。
浓度应精确至0.0001mol/L。
平行试验不得少于8次,两人各做4次,每人4次平行测定结果的极差与平均值之比不得大于0.2%,两人测定结果平均值之差不得大于0.2%。结果取平均值。
(3)测定:称取试样5g于500mL克氏烧瓶中,精确至0.001g。加瓷环2~3个,加缓冲溶液75mL,将试样冲至瓶底,立即将瓶连接于水蒸气蒸馏装置,用内含15mL0.1mol/L盐酸标准滴定溶液的1000mL三角瓶作接收器,冷凝管末端浸入盐酸标准滴定溶液中。开始蒸馏,馏出液温度不应显着高于室温,蒸馏过程中应保持克氏烧瓶中液体体积恒定。必要时可适当加热。馏出液达800~850mL时,取下三角瓶,同时用水冲洗冷凝管末端,停止蒸馏。向馏出液中加入8~10滴甲基红-次甲基蓝混合指示剂,用0.1mol/L氢氧化钠标准滴定溶液滴定溶液至浅灰色。同时做空白实验,并加以校正。
5.计算
按下式计算总挥发碱(以氨计)质量分数:

式中 wh——以NH3表示的总挥发碱的质量分数,%;
V0——空白试验耗用的盐酸标准滴定溶液的体积,mL;
V1——加入盐酸标准滴定溶液的体积,mL;
V2——滴定耗用的氢氧化钠标准滴定溶液的体积,mL;
c1——盐酸标准滴定溶液的浓度,mol/L;
c2——氢氧化钠标准滴定溶液的浓度,mol/L;
m——试样质量,g;
w——试样的含水率,%;
17——NH3的摩尔质量,g/mol。
以2次平行测定的平均值作为测定结果,精确至0.01%。2次平行测定结果的相对偏差不应大于6.0%。
(十一)烟草中去甲基烟碱的测定
1.紫外吸收光谱法
(1)原理:利用亚硝酸钠与第二胺基生物碱(主要为去甲基烟碱)的反应,从水蒸气蒸馏液中把烟碱和去甲基烟碱分离开后,测定混合物以及只含有烟碱的试样的紫外吸收光谱,并对两者进行定量。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①氯化钠。
②20%氢氧化钠溶液。
③0.5mol/L盐酸溶液。
④盐酸(1∶4)。
⑤6%硅钨酸(4H2O·SiO2·12WO3·22H2O)溶液。
⑥30%醋酸(mL/mL)溶液。
⑦亚硝酸钠。
⑧1%的酚酞溶液。
⑨4%氢氧化钠溶液。
(3)仪器:
蒸汽蒸馏装置。
具有波幅为2nm以下性能的分光光度计。
(4)方法:
①准确称取2~3g的试样,放入水蒸气蒸馏装置的蒸馏瓶中。
②加入约10g的氯化钠和约6mL20%氢氧化钠溶液,进行蒸汽蒸馏。
③将馏出液收容在20mL0.5mol/L盐酸溶液中。
④蒸馏后期的馏出液接到试管中,加入盐酸(1∶4)和6%硅钨酸溶液各数滴,如仍有生物碱馏出则呈白色混浊物,不再出现白色混浊则生物碱已全部馏出。
⑤将馏出液放入200mL的容量瓶中,加水定容至200mL。
⑥准确吸取上述定容后的蒸馏液10mL放入蒸馏瓶中,滴入酚酞作指示剂。用4%氢氧化钠溶液中和后,加入30%醋酸溶液2mL和亚硝酸钠0.5g,在室温下放置20min,试样中去甲基烟碱与亚硝酸钠反应,生成用蒸汽蒸馏难于得到的亚硝基化合物。
⑦在蒸馏瓶中加入约10g的氯化钠和约6mL20%氢氧化钠溶液,再进行蒸汽蒸馏。
⑧将馏出液收容在10mL0.5mol/L盐酸溶液中。
⑨蒸馏后期的馏出液接收到试管内,加入盐酸(1∶4)和6%硅钨酸溶液各数滴,如有烟碱馏出则呈白色混浊物,否则烟碱已全部馏出。
⑩当确认烟碱已全部馏出后,停止蒸馏,将蒸馏液移入100mL容量瓶中,用水定容。
⑾另外从方法⑤的蒸馏液中准确称取10mL放入100mL容量瓶中,加入0.5mol/L盐酸溶液9mL,用水定容。
⑿将方法⑩、⑾稀释后的蒸馏液放入分光光度计1cm厚的比色皿中,测定其236nm、259nm、282nm的吸光度。
(5)计算:
①方法①所得试样(烟碱和去甲基烟碱的混合物)的计算按下式计算:
A′259=1.059[A259-1/2(A236+A282)]
式中 A′259——修正后的烟碱+去甲基烟碱在259nm处的吸光度;
A236——236nm处的吸光度;
A259——259nm处的吸光度;
A282——282nm处的吸光度。
②方法⑩所得试样(只含烟碱)的计算:
A′259=1.059[A259—1/2(A236+A282)]
式中 A′259——修正后的烟碱在259nm处的吸光度;
A236——236nm处的吸光度;
A259——259nm处的吸光度;
A282——282nm处的吸光度。
③方法⑩所得试样中的烟碱含量按下式计算:
m′=A′259/343×b
式中 m′——100mL被测液中的烟碱含量,g;
A′259——修正后的烟碱在259nm处的吸光度;
343——烟碱的吸光系数×10;
b——比色皿的厚度,cm。
④方法①所得试样的去甲基烟碱的含量按下式计算:

式中 A″259——修正后的去甲基烟碱在259nm处的吸光度;
 ——修正后的烟碱+去甲基烟碱在259nm处的吸光度;
——修正后的烟碱+去甲基烟碱在259nm处的吸光度;
A′259——修正后的烟碱在259nm处的吸光度。
m”=A″259/387×b
式中 m″——100mL被测液中的去甲基烟碱含量,g;
A″259——修正后的去甲基烟碱在259nm处的吸光度;
387——去甲基烟碱的吸光系数×10;
b——比色皿的厚度,cm。
⑤烟碱、去甲基烟碱的含量w烟碱、w去甲基烟碱,以%表示,按下式计算:

式中 m——试样质量,g。
2.气相色谱法
(1)原理:将试样用含有一量内标物的0.5mol/L盐酸溶液萃取后,在碱性条件下,把去甲基烟碱和内标溶液转溶到三氯甲烷层中,用气相色谱仪对去甲基烟碱定量。
(2)仪器、设备及试剂:
①气相色谱仪,配有氢火焰离子检测器及数据处理装置。
②分析天平:感量为万分之一克。
③自动吸量管。
④注射器。
⑤色谱柱:内径在2~4mm之间,长度在1.5~2m之间,不锈钢柱。
⑥载气:氮气。
⑦标准筛:80~100目/25.4mm2。
⑧固定相:PEG-20M涂布(质量分数20%)的Chomosorb W-AW,60~80目/25.4mm2。
⑨去甲基烟碱·异喹啉标准溶液:准确称取0.5g和1.0g去甲基烟碱,分别准确加入2g异喹啉,使其溶解在约230mL的盐酸(1∶4)中,用水稀释到1L,放入棕色瓶内在冰箱中保存。
⑩0.5mol/L盐酸溶液(含有内标物异喹啉):准确称取20g异喹啉,用2.3L盐酸(1∶4)溶解后稀释,定容为10L。
⑾特级氯仿。
⑿30%氢氧化钠溶液。
⒀瓦特曼1PS滤纸。
(3)方法:色谱工作条件
柱温:170~190℃。
进样口温度:250℃。
载气:氮气(流量60~80mL/min)。
进样量:3μL。
①色谱柱的制备:预先准备好用PEG-20M涂布(质量比20%)的Chomosorb W-AW(60~80目/25.4mm2)担体。称取担体质量1%的氢氧化钠,将上述担体加入氢氧化钠甲醇溶液,再用旋转蒸发器减压干燥,即为分析烟碱用的填充剂。
依次用稀盐酸、水和氢氧化钠(1+100)清洗不锈钢柱的内壁,然后注入100mL约30%的氢氧化钠溶液,再用甲醇、丙酮和乙醚依次除去支子中的含水率,干燥后加入填充剂。
②准确称取约1g试样放入50mL具塞三角瓶中,用自动吸液管准确吸取一定量(10~20mL)0.5mol/L盐酸溶液(含有内标物)加入三角瓶中,盖好塞子,振动萃取5min。
③加入30%的氢氧化钠约2mL和氯仿约10mL,轻轻摇振后,去甲基烟碱和内标物即转溶到氯仿层中。
④将烧瓶中的内容物转移到瓦特曼滤纸上,即可得到氯仿的滤液。
⑤用微量注射器吸取3μL滤液,注入汽化室。
⑥分析试样开始之前以及每测定10~20个试样后,需在相同的条件下,对两种浓度的去甲基烟碱-异喹啉标准溶液进行测定,然后求出去甲基烟碱于异喹啉的峰面积比或峰高比。
(4)计算:根据2种去甲基烟碱-异喹啉标准溶液色谱图得到:


式中 A1——去甲基烟碱的面积或峰高;
A2——异喹啉的面积或峰高;
m1——去甲基烟碱的质量,g;
m2——异喹啉的质量,g。
由此求出面积或峰高比和质量比的关系:
Y=ax+b
式中 a、b——常数。
去甲基烟碱的含量w甲基烟碱,以%表示,按下式计算:

式中 A1——去甲基烟碱的面积或峰高;
A2——异喹啉的面积或峰高;
m3——在萃取的过程中使用的10~20mL0.5mol/L盐酸中异喹啉的质量,g;
m——试样的质量,g。
(十二)烟草中甘油、丙撑二醇和三甘醇的测定——气相色谱法
1.原理
试样用无水甲醇萃取,得到的萃取液直接进行气相色谱分析。
2.试剂和材料
所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
(1)无水甲醇。
(2)1,3丁二醇。
(3)内标物1,3丁二醇标准溶液:准确称取20g1,3丁二醇,放入100mL容量瓶中,用无水甲醇稀释定容。
(4)萃取溶液:准确吸取20mL内标物1,3丁二醇标准溶液,用无水甲醇稀释到2L。
(5)甘油标准溶液:准确称取10g甘油,放入100mL容量瓶中,用萃取溶液稀释并定容。
(6)丙撑二醇标准溶液:准确称取5g丙撑二醇,放入100mL容量瓶中,用萃取溶液稀释并定容。
(7)三甘醇标准溶液:准确称取5g三甘醇,放入100mL容量瓶中,用萃取溶液稀释并定容。
(8)校正曲线用标准溶液:取四个100mL容量瓶,分别加入1.0mL、2.0mL、3.0mL、4.0mL甘油标准溶液、丙撑二醇标准溶液和三甘醇标准溶液。用萃取溶液稀释定容。
3.方法
(1)准确称取10g试样放置于250mL标准磨口锥形瓶中,加入100mL萃取溶液,盖上盖子,机械振荡30min。静置过夜。
(2)用微量注射器分别吸取校正曲线用标准溶液各30μL注入气相色谱仪进行气相色谱分析。根据峰高比和质量制作校正曲线。
(3)气相色谱仪的条件:气相色谱仪,配有热W丝检测器及数据处理装置。
检测器桥电流:140mA。
色谱柱:0.35mmi.d.×1m铜管,填充剂为5%Carbowax 20M-对酞酸涂渍的60~80目Chomosorb GAW-DMCS。
填充剂的制备:将30gChomosorb置于500mL标准磨口圆底烧瓶中,加入150mL氯仿与1.5gCarbowax 20mol/L-对酞酸的混合溶液,调成浆状。在旋转蒸发器内真空条件下除去氯仿,然后在室温下干燥过夜。新制备的填充剂要在240℃条件下老化2h,然后在分析试样前先注入30μL烟草试样萃取液3次。
检测器温度:280℃。
进样口温度:265℃。
柱温:90~240℃(15℃/min)。
载气:He,60mL/min。
试样注入量:30μL。
(4)测定试样萃取液,根据试样萃取液中甘油、丙撑二醇和三甘醇对1,3丁二醇的峰高比,求出试样中甘油、丙撑二醇和三甘醇的含量。
(十三)烟草中挥发性有机酸(甲酸、乙酸)的测定
1.原理
在试样中加入少量的磷酸溶液,使烟草中的甲酸和乙酸生成相应的盐游离后,用乙醇萃取,将这些酸转换成碱金属盐后干固,再在浓硫酸存在的条件下丁酯后用气相色谱法分析。
2.试剂和材料
所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
(1)1.3mol/LH3PO4。
(2)戊酸水溶液(内标):将约300mg戊酸放入200mL容量瓶中用纯水定容到刻度。
(3)1%碳酸氢钠水溶液。
(4)正丁醇。
(5)浓硫酸。
(6)无水硫酸钠。
(7)正己烷。
(8)乙醚。
(9)甲酸和乙酸水溶液(制作校正曲线用):
①准确称取无水甲酸钠和无水乙酸钠各100mg放入50mL容量瓶中,用戊酸内标溶液定容为50mL。
②取25mL溶液1放入50mL容量瓶中,用戊酸内标溶液定容为50mL。
③取25mL溶液2放入50mL容量瓶中,用戊酸内标溶液定容为50mL。
3.仪器
气相色谱仪,配有氢火焰离子检测器。
旋转式蒸发仪。
恒温水浴锅。
4.方法
(1)准确称取2g的试样,放入50mL的烧杯中,加入4mol/L磷酸2mL和戊酸内标溶液1mL,用刮勺充分搅混。
(2)将混合物移入200mL的分液漏斗中,加入100mL乙醚和15g无水硫酸钠,用摇床摇振10min。
(3)将乙醚溶液用滤纸过滤,滤液移入100mL的分液漏斗中,加入10mL1%碳酸氢钠水溶液,摇振5min。
(4)将水层移入约30mL梨型烧瓶中,用50℃以下的水浴在旋转式蒸发仪上减压干燥。
(5)在干燥后的固体物内加入2mL正丁醇,0.25mL浓硫酸,2g无水硫酸钠。装上回流冷凝器,在硅油浴上用100℃加热15min。
(6)反应后的溶液放至室温,加0.5mL正己烷和5mL水,摇振10s。
(7)将正己烷层用安全移液管移入小试管内,加入约正己烷3倍的水,摇振10s。
(8)将正己烷层移入另一个小试管内,加入少量的无水硫酸钠,作为气相色谱分析的测试样。
(9)气相色谱分析的条件:
柱子:PEG1500,0.25mmi.d×50mm。
温度:60℃,恒温。
载气:N,0.5~0.8mL/min。
进样量:1~2mL。
(10)测定色谱图中甲酸、乙酸和戊酸丁酯的峰高。
(11)按同样的实验操作,测定加1mL校正曲线用有机酸溶液的试样,根据所得到的峰高与不加试样的校正曲线用有机酸溶液的峰高差,求得戊酸丁酯的峰高,再根据它与甲酸丁酯及乙酸丁酯的峰高比,制作校正曲线。
(12)同一个试样要分析2次以上。
5.计算
(1)制作校正曲线所用的标准溶液中的甲酸和乙酸含量:

式中 A——1mL试剂的溶液中甲酸的含量,mg/mL;
B——1mL试剂的溶液中乙酸的含量,mg/mL;
m1——称取的甲酸钠质量,mg;
m2——称取的乙酸钠质量,mg。
(2)校正曲线:上述的每1mL各标准溶液中有机酸的含量和由分析得到的各种有机酸与内标物质峰高比,分别在坐标纸上做图,制成校正曲线,确定两者之间成直线关系的范围,由此求出它们的关系式。
Y=ax+b
式中 Y——有机酸的含量;
x——内标物质和各种有机酸的峰高比;
a,b——由坐标图求得的常数。
(3)试样中的有机酸量:从上式求出试样中的甲酸和乙酸的含量,再按下式计算试样中的甲酸和乙酸的质量分数,以%表示:

式中 m1——甲酸的含量,mg;
m2——乙酸的含量,mg;
m——试样的质量,g。
(十四)烟草中非挥发性有机酸(草酸、苹果酸、柠檬酸)的测定
1.原理
在试样中加入硫酸-甲醇,进行萃取,同时使有机酸甲酯化,再用二氯甲烷萃取生成的酯,然后进行气相色谱分析。
2.试剂和材料
所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
(1)12.5%的硫酸-甲醇溶液;
(2)戊二酸-甲醇溶液(内标):准确称取1000~1100mg戊二酸放入1000mL容量瓶中,用甲醇定容到刻度;
(3)有机酸-甲醇溶液(制作标准曲线用):在三个100mg容量瓶中,分别加入准确称量的草酸、苹果酸、柠檬酸50mg、500mg、500mg。加入戊二酸-甲醇溶液,并放在水浴上加热使有机酸溶解,冷却至室温再用戊二酸-甲醇溶液定容到刻度;
(4)二氯甲烷。
(5)无水硫酸钠。
3.仪器
气相色谱仪,配有氢火焰离子检测器。
旋转式蒸发仪。
恒温水浴锅。
4.方法
(1)准确称取1g的试样,放入100mL的茄型烧瓶中,准确加入10mL戊二酸-甲醇溶液和12.5%的硫酸-甲醇溶液40mL,在水浴水加热后,用滤纸过滤。
(2)取25mL滤液和50mL水放入100mL的分液漏斗中,每次用5mL二氯甲烷萃取,共萃取2次。
(3)在一个分液漏斗里放上脱脂棉,其上放约3g无水硫酸钠。将萃取后的二氯甲烷溶液过滤,此溶液作为气相色谱分析的试样。
(4)气相色谱分析的条件:
柱子:PEG20M,0.25mmi.d.×30mm。
温度:100~200℃,5℃/min。
载气:He,1mL/min。
进样量:1L。
(5)测定色谱图中的峰高。
(6)按同样的实验操作,测定加10mL校正曲线用有机酸溶液的试样。
(7)同一个试样要分析2次以上。
5.计算
(1)用校正曲线法的常规方法计算。
(2)试样中的有机酸量按下式计算:

式中 w有机酸——有机酸的质量分数,%;
m′——有机酸的含量,mg;
m——试样的质量,g。
(十五)烟草中淀粉的测定
1.高氯酸萃取法
(1)原理:用高氯酸萃取淀粉,再用碘与其生成复合体,从高氯酸中分离后用盐酸加含水率解,对葡萄糖定量。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①30%高氯酸溶液。
②碘-碘化钾溶液:将7.5g碘和7.5g碘化钾溶解在水中,并定容为250mL,然后过滤。
③乙醇-氯化钠溶液:将350mL乙醇,80mL水和20%的氯化钠混合后,稀释到500mL。
④乙醇-氢氧化钠溶液:将350mL乙醇,100mL水和25mL5mol/L氢氧化钠混合后,定容为250mL,然后过滤。
⑤1mol/L盐酸溶液。
⑥20%的氯化钠溶液。
⑦0.05%酚红指示剂:将50mg酚红溶解在100mL乙醇中。
⑧0.5mol/L氢氧化钠溶液。
⑨0.05mol/L草酸溶液。
⑩索莫季试剂:将30g酒石酸钾钠(KNaC4H4O6·4H2O)和30g无水碳酸钠溶解在200mL热水中,加入40mL1mol/L的氢氧化钠溶液,再加用30mL水溶解的8g硫酸铜(CuSO4·5H2O),加热15min赶出溶液中的空气。另取180g无水硫酸钠溶解在约500mL热水中,加热排出溶液中的空气。冷却后将上述两溶液移入1L的容量瓶中。另用约10mL的水溶解8g碘化钾,并加适量的碘酸钾,加入上述溶液,冷却后加水到1L。
⑾中性醋酸铅溶液:将36g中性醋酸铅[Pb(CH3COO)2·3H2O]溶解在95mL水中。
⑿无水草酸钠。
⒀2.5%盐酸溶液。
⒁1mol/L氢氧化钠溶液。
⒂1mol/L硫酸溶液。
⒃0.05mol/L硫代硫酸钠溶液:将25g硫代硫酸钠溶解于1L水中,加热并使其沸腾5min,趁热装入褐色瓶中,于低温避光处保存。该溶液的系数按下述方法求出。
精确称取经130℃干燥1h的特级碘酸钾0.15g,放入200mL的三角瓶中,加水100mL,在加2g碘化钾和1mol/L硫酸溶液15mL,用硫代硫酸钠溶液滴定游离碘,当黄色基本消失时,加入淀粉指示剂。紫色消失时为滴定终点。系数F用下式求出:

式中 m——碘酸钾的质量,mg;
T——滴定值,mL。
⒄0.0025mol/L硫代硫酸钠溶液:由于该浓度的溶液不稳定,应先配制0.05mol/L的溶液,准确标定后存放起来,使用前再稀释到0.0025mol/L。
⒅淀粉指示剂:将1g可溶性淀粉放入20mL冷水中搅成糊状,再倒入140mL沸水中,搅匀煮沸5min,加20g氯化钠,冷却后用水定容到200mL。
(3)方法:
①准确称取0.5~1g试样放置于50mL容量的耐热试管中。
②加入12mL水,在水浴上加热15min,使淀粉糊化后,冷却至室温。
③再放到22~25℃的水浴上保温,一边搅拌,一边迅速加入9mL高氯酸。
④用粗搅拌棒研磨试管底部的试样,萃取15~20min,将搅拌棒用少量30%的高氯酸溶液清洗,将洗液合并入萃取液中,然后进行离心分离。
⑤分离后将上部清液移入100mL的容量瓶中,残渣再按方法②、③的比例分别加入水和高氯酸,进行同样的研磨萃取。反复进行这一操作,将萃取液合并置于100mL的容量瓶中,用水定容,盖上盖后充分摇混。
⑥定容后的萃取液再进行离心分离。如有必要,上部清液可用玻璃棉过滤。
⑦准确吸取5~10mL滤液,放入耐热玻璃离心试管中,稀释到10mL。再加入5mL20%的氯化钠溶液,以及2mL碘-碘化钾溶液,混合后至少放置20min以上。
⑧经离心机分离后,用安全移液管将上部清液吸出不要,留下沉淀。
⑨在沉淀中加入5mL乙醇-氯化钠溶液,使之成为悬浊液以清洗沉淀,再用离心法分离。
⑩再在方法⑨的沉淀中加5mL乙醇-氯化钠溶液,振摇至蓝色为止,再用离心法分离游离的淀粉。
⑾沉淀出的淀粉用5mL乙醇-氯化钠溶液清洗并离心分离,然后加入5mL0.1mol/L的盐酸溶液,盖上离心管盖子,在水浴上水解2h30min。
⑿水解后放冷,以酚红作指示剂,用0.5mol/L的氢氧化钠溶液中和后,再用0.05mol/L的草酸溶液消色。根据淀粉量稀释定容。
⒀吸取一定量的上述溶液于25mm×200mm的试管中,加索莫季试剂5mL。
⒁另取一同样试管,分别加入水和试剂各5mL,作为空白对照。
⒂试管口加玻璃球盖,将数支试管放入金属框中,然后在沸腾水浴上加热15min。
⒃加热完毕后立即用自来水冷却,再加入1mol/L硫酸溶液2mL。硫酸要迅速加入,同时充分摇混使溶液一下成为酸性。
⒄用0.0025mol/L硫代硫酸钠溶液滴定游离碘,当黄色基本消失时,加入淀粉指示剂。紫色消失时为滴定终点。
(4)计算:
按下式计算淀粉的含量w淀粉,以%表示:

式中 m′——每5mL用莫索季法得到的水解液中糖的含量,mg;
V——水解液定容后的容量,mL;
m——试样的质量,g。
2.糖量分析仪法
(1)原理:试样先用80%的乙醇溶液萃取,残渣用盐酸水解后,用糖量分析仪定量水解液中的葡萄糖量。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①80%乙醇。
②滑石粉悬浊液:在200g滑石粉中加入约1L的水,充分搅拌。在悬浊状态下使用。
③25%盐酸溶液。
④20%氢氧化钠溶液。
⑤2%氢氧化钠溶液。
⑥强酸性离子交换树脂(H型):用盐酸(1+6)充分洗涤该树脂,然后再用水充分洗涤使洗液呈中性为止。
⑦强碱性离子交换树脂(OH型):用5%氢氧化钠溶液充分洗涤该树脂,然后再用水充分洗涤使洗液呈中性为止。
⑧葡萄糖标准溶液:将葡萄糖在100℃干燥1h后,精确称取2.000g,溶解在0.25%苯甲酸溶液中,准确定容为1L。
(3)仪器:糖量自动分析仪。
(4)方法:
①准确称取约1g试样于200mL三角瓶中,加入约80mL80%的乙醇,装好回流冷却装置,在水浴上加热萃取1.5h。
②将滑石粉悬浊液10mL放在布氏漏斗上吸滤,然后再把冷却后的萃取液在该漏斗上吸滤。残渣用80%的乙醇洗涤至无色。
③残渣及滑石一起移入200mL三角瓶中,在60℃下干燥,直到无乙醇味为止。再加入70mL水和7mL25%的盐酸溶液。装好回流冷却装置,在水浴上加热水解2.5h。
④将水解物减压过滤在200mL的烧杯中,残渣用温水洗净。合并滤液和洗液。
⑤冷却后用20%及2%氢氧化钠溶液中和,再过滤在200mL的烧杯中,加水稀释到刻度。
⑥量取上述溶液80mL,放入100mL三角瓶中,加入强酸性离子交换树脂约200mg和强碱性离子交换树脂200mg,盖上盖子充分摇混。
⑦放置10~20min后过滤,滤液即用于糖量自动分析仪测定。
⑧接通分析仪的电源2h后,将电极插入传感器。
⑨用仪器带的缓冲剂配制缓冲液,放入缓冲液贮瓶。然后将开关拨到standby侧,按清除键30s。
⑩将开关拨向Run侧,按清除键,Zero/inject灯亮后,用微量注射器吸取葡萄糖标准溶液,从进样口注入,放置2min。
⑾按清除键,Zero/inject灯亮后,调整零点微调旋钮,使浓度表示(mg/100mL)为0。
⑿注入葡萄糖标准溶液之后,按定标键,在Ready灯亮的同时,用定标微调旋钮将浓度指示调整到200。
⒀重复方法①、②,使Ready灯亮后约40s,浓度指示稳定在198~202范围内。
⒁按清除键,Zero/inject灯亮后,用零点微调旋钮调整浓度指示为0。
⒂吸取方法⑦的滤液注入仪器,Ready灯亮后记录下浓度值。
⒃测定结束后按清除键,Zero/iniect灯亮,将开关拨向Standby侧,把电极插头拔掉,切断电源。
(5)计算:
按下式计算淀粉的含量w淀粉,以%表示:

式中 ρ——浓度读数,mg/100mL;
m——试样的质量,g。
(十六)烟草中淀粉及糊精的测定
1.加入80%乙醇萃取法
(1)原理:在试样中加入80%乙醇溶液,萃取糖,残渣用盐酸水解后,经除去蛋白质处理制成糖液,对葡萄糖定量。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①80%乙醇溶液。
②索莫季试剂:将30g酒石酸钾钠(KNaC4H4O6·4H2O)和30g无水碳酸钠溶解在200mL热水中,加入40mL1mol/L的氢氧化钠溶液,再加用30mL水溶解的8g硫酸铜(CuSO4·5H2O),加热15min赶出溶液中的空气。另取180g无水硫酸钠溶解在约500mL热水中,加热排出溶液中的空气。冷却后将上述两溶液移入1L的容量瓶中。另用约10mL的水溶解8g碘化钾,并加适量的碘酸钾,加入上述溶液,冷却后加水到1L。
③中性醋酸铅溶液:将36g中性醋酸铅[Pb(CH3COO)2·3H2O]溶解在95mL水中。
④无水草酸钠。
⑤25%盐酸溶液。
(3)方法:
①准确称取1.5g试样(晾晒烟及白肋烟为1g),放置于200mL圆底烧瓶或三角烧瓶中,加80%乙醇溶液约100mL,装上回流冷凝装置,在水浴上加热萃取1h。
②冷却后取上层清液,残渣再加100mL乙醇溶液,用上述的方法萃取,再用布氏漏斗过滤,并用80%乙醇溶液清洗残渣数次,至滤液无色为止。
③洗后残渣在60℃下干燥,使其完全消除乙醇的气味。
④将干燥后的残渣放置于200mL的三角烧瓶中,加水70mL,再加7mL盐酸溶液,装上回流冷凝装置,在水浴上水解2.5h。
⑤将分解物过滤到200mL容量瓶中,并用温水清洗残渣,洗液和滤液合并到一起。
⑥冷却后用1mol/L氢氧化钠溶液中和,再加水到200mL。
⑦准确吸取上述糖液10mL(晾晒烟及白肋烟为25mL),放置于100mL容量瓶中,加中性醋酸铅溶液,直到不出现沉淀为止。充分振摇后加水至100mL。
⑧用干燥的滤纸过滤,在滤液中加无水草酸钠,直到不出现沉淀为止。充分搅拌后放置1.5~2h,再用干燥滤纸过滤。
⑨准确吸取上述滤液5mL于25mm×200mm的试管中,加索莫季试剂5mL。
⑩另取一同样试管,分别加入水和试剂各5mL,作为空白对照。
⑾试管口加玻璃球盖,将数支试管放入金属框中,然后在沸腾水浴上加热15min。
⑿加热完毕后立即用自来水冷却,再加入1mol/L硫酸溶液2mL。硫酸要迅速加入,同时充分摇混使溶液一下成为酸性。
⒀用0.0025mol/L硫代硫酸钠溶液滴定游离碘,当黄色基本消失时,加入淀粉指示剂。紫色消失时为滴定终点。
(4)计算:
按下式计算烤烟的淀粉及糊精:

按下式计算晒烟或白肋烟的淀粉及糊精:

式中 w1——淀粉+糊精的质量分数,%;
m′——每5mL试样溶液用莫索季法得到的水解液中的含糖量,mg;
m——试样的质量,g。
2.自动分析仪法
(1)原理:在试样中加入80%乙醇溶液,萃取糖,残渣用盐酸水解后,放入自动分析仪除去蛋白质,测定还原能力,对葡萄糖进行定量。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①30%Brij35[聚氧化乙烯(23)月桂醚]溶液。
②0.25%硫酸锌和0.25%硫酸铝钾[KAl(SO)4·12H2O]的混合溶液:将5g硫酸锌(ZnSO4·7H2O)和5g特级硫酸铝钾溶解于水中,定容为2L。再加入1mLBrij35。
③0.033mol/L氢氧化钠溶液:将2.7g氢氧化钠溶解在煮沸后除去二氧化碳的软化水中,定容为2L。再加入1mL30%Brij35。
④1mol/L氢氧化钠溶液:将40g氢氧化钠溶解在煮沸后除去二氧化碳的软化水中,定容为1L。再加入0.5mL30%Bri.j35。
⑤0.046%碱性铁氰化钾溶液:将0.92g特级铁氰化钾[赤血盐K3Fe(CN)6]溶解于水中后,加入16g氢氧化钠,溶解后用水定容为2L。再加入1mL30%Brij35。
⑥1%氰化钾溶液:将20g氰化钾溶解于软化水中,定容为2L。再加入1mL30%Brij35。
⑦0.25%苯甲酸溶液:将5g苯甲酸加温溶解于软化水中,定容为2L。
⑧葡萄糖标准原液:将葡萄糖在100℃干燥1h后,精确称取10.000g,溶解在0.25%苯甲酸溶液中,准确定容为1L。
⑨葡萄糖标准溶液:分别吸取原液2mL、5mL、10mL、15mL,用0.25%苯甲酸溶液准确定容为100mL。该标准溶液在100mL中含有葡萄糖分别为20mg、50mg、100mg、150mg。
(3)仪器:自动分析仪。
(4)方法:
①准确称取约5g试样,放入200mL的茄型烧瓶中,加入100mL80%的乙醇,装好回流冷却装置,在沸水浴上加热萃取1h。
②放冷后去掉上部清液,再在残渣中加入100mL80%的乙醇,装好回流冷却装置,在沸水浴上加热萃取1h。用布氏漏斗过滤,并用80%的乙醇冲洗至滤液接近无色为止。
③将残渣在60℃下干燥,使其完全消除乙醇的气味。
④将干燥后的残渣放入200mL的三角烧瓶中,加水70mL,再加7mL2.5%盐酸溶液,装上回流冷凝装置,在沸水浴上水解2.5h。
⑤减压过滤,再将残渣用湿水洗净,将洗液和滤液合并到一起。
⑥冷却后用1mol/L氢氧化钠溶液中和,浓缩冷却后,准确定量为50mL。
⑦将自动分析仪的各试剂输管装入软化水中,关闭取样器软化水阀门,打开泵、比色计的开关,冷却器内通入水。
⑧分别将配制好放置30min以后的0.25%硫酸锌和0.25%硫酸铝钾的混合溶液、0.033mol/L氢氧化钠溶液、0.3mol/L盐酸溶液、0.046%碱性铁氰化钾溶液、1mol/L氢氧化钠溶液、1%氰化钾溶液用泵吸入。
⑨在取样器上同时准备好葡萄糖标准溶液和方法①的滤液。
⑩打开过滤装置、取样器、记录仪的开关。
⑾取样结束后,关掉取样器。
⑿10min后将0.25%硫酸锌和0.25%硫酸铝钾的混合溶液、0.033mol/L氢氧化钠溶液的输液管内换成软化水,15min后在0.3mol/L盐酸溶液输液管内换成软化水。
⒀在装有0.033mol/L氢氧化钠溶液的输液管用0.3mol/L盐酸溶液冲1min,再放入软化水。该酸性溶液不能进入泵中,应在过滤液开始取样之前拿掉。然后倒掉管中的0.3mo1/L盐酸,这时过滤液管用软化水冲洗。
⒁关掉过滤装置。
⒂根据记录,拔掉最后一个试样前两个盛有0.046%碱性铁氰化钾溶液的输液管,放入软化水中。最后一个试样输液管仍按原状不动。
⒃记录纸上记录结束以后,关闭记录仪、比色计。15min后关掉泵以及取样器的软化水和冷却器用水。
⒄根据记录仪上标准溶液的吸光度制作标准曲线,由此求出100mL试样溶液中含的毫克数。
(5)计算:按下式计算淀粉及糊精的含量w1,以%表示:

式中 m′——100mL试样溶液中的含糖量,mg;
m——试样的质量,g。
3.气相色谱法
(1)原理:试样用80%乙醇溶液萃取后,残渣用盐酸加含水率解,将得到的葡萄糖甲硅烷基化后进行气相色谱分析。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①80%乙醇溶液。
②甲硅烷基化试剂TMS-PZ。
③内标物山梨糖醇液:准确称取约400mg山梨糖醇,放入100mL容量瓶中,加水定容为100mL。
④约10mol/L的NaOH溶液。
⑤25%盐酸溶液。
⑥糖类校正曲线用溶液:
a.准确称取240mg无水葡萄糖放入100mL容量瓶中,加水定容为100mL;
b.准确吸取50mLa溶液放入100mL容量瓶中,加水定容为100mL;
c.准确吸取10mLa溶液放入100mL容量瓶中,加水定容为100mL。
(3)方法:
①准确称取1g试样放置于300mL茄型烧瓶或三角烧瓶中,约加150mL80%乙醇,装上回流冷凝器在沸水浴上加热1h。冷却后,加山梨糖醇水溶液(烤烟5mL,其他类型烟1mL)用同山式漏斗(No.5A滤纸)过滤。再用40mL80%乙醇洗净。
②用35mL水将残渣冲入200mL的茄型烧瓶中,加3.5mL25%盐酸,装上回流冷凝器用电热罩加热2.5h。冷却后用10mol/L的氢氧化钠溶液将pH值调节到7,加山梨糖醇水溶液(烤烟5mL,其他类型烟1mL)过滤。
③取一定量(烤烟30μL,其他类型烟50μL)滤液放入2mL的试管中,在干燥器中减压干燥后,用微量注射器加40μLTMS-PZ,放置约15min后进行气相色谱仪分析。
④气相色谱仪的条件:
气相色谱仪,配有氢火焰离子检测器及数据处理装置。
色谱柱:OV 101,0.25mmi.d.×30m玻璃毛细管。
温度:150~280℃(3℃/min)。
载气:He,0.9mL/min。
试样注入量:1μL,感度:10×8。
⑤测定色谱图中葡萄糖的峰高。
⑥分别吸取校正曲线用溶液各10mL放入三角烧瓶中,再加1mL山梨糖醇内标液,混合后各取20μL分别放入2mL的试管中,在干燥器中减压干固后,用微量注射器分别加入30μLTMS-PZ,放置约15min后进行气相色谱仪分析。根据峰高比和质量制作校正曲线。
(4)计算:根据校正曲线计算试样中葡萄糖的含量,其值乘以0.9即为淀粉和糊精的含量。
(十七)烟草中多酚类物质(绿原酸、芸香苷)的测定
1.高效液相色谱法
(1)原理:将试样用正己烷萃取,脱脂后试样再用80%的甲醇处理,萃取出多酚类。最后用高效液相色谱仪分析。
(2)试剂:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①正己烷(也可使用正戊烷或石油醚)。
②80%的甲醇水溶液。
③0.1的磷酸二氢钾(KH2PO4)水溶液:将6.8g的磷酸二氢钾(KH2PO4)用水溶解后定容到1L。
④15%的甲醇溶液:将甲醇与方法③的磷酸二氢钾水溶液以15∶85(体积分数)混合即可。
⑤35%的甲醇溶液:甲醇与方法③的磷酸二氢钾水溶液以35∶65(体积分数)混合即可。
⑥七叶亭-甲醇溶液(绿原酸用的内部标准溶液):用丙酮将400mg的七叶亭溶解后,再用80%的甲醇水溶液定容到100mL。
⑦香豆素-甲醇溶液(芸香苷用的内部标准溶液):再用80%的甲醇水溶液将400mg香豆素溶解并定容到100mL。
⑧绿原酸-甲醇溶液:
a.将10.0mg的绿原酸放入10mL的容量瓶,用80%的甲醇水溶液定容到10mL;
b.取5mL溶液a,再用80%的甲醇水溶液定容到10mL;
c.取2mL溶液a,加80%的甲醇水溶液定容到10mL。
⑨芸香苷-甲醇溶液:
a.取40mg芸香苷放入10mL的容量瓶中,用80%的甲醇水溶液定容到10mL;
b.取5mL溶液a,用80%的甲醇水溶液定容到10mL;
c.取2mL溶液a,加80%的甲醇水溶液定容到10mL。
(3)仪器、设备:高效液相色谱仪:
①柱子: C18,4mmni.d.×40cm。
C18,4mmni.d.×40cm。
②展开溶液:15%的甲醇溶液(绿原酸用),35%的甲醇溶液(芸香苷用)。
③流速:1.4mL/min。
④压力:70~90kg/cm2。
⑤检测器:328nm可见光区的吸光检测器。
⑥灵敏度:0.02a.m.f.s.。
⑦进样量:1~3μL。
(4)方法:
①准确称取约2g的烟叶粉末放入索式萃取器中,用正己烷(或正戊烷或石油醚)萃取2h。
②将圆筒滤纸中的脱脂试样用60mL80%的甲醇水溶液转移到100mL容量的茄型烧瓶中,在水浴中回流加热30min。
③测定绿原酸时,在萃取溶液中加5mL七叶亭-甲醇溶液;测定芸香苷时改加5mL香豆素-甲醇溶液,再用滤纸过滤即可做高速液相色谱仪测试。
④各分析均以多酚类内标物的峰面积比和浓度比做出标准曲线,再求出试样中多酚类的含量。
⑤制作校正曲线是用绿原酸标准溶液,即试剂(2)之8中a、b、c、溶液各2mL,分别不加0.1mL七叶亭-甲醇溶液,混合后进行分析,同样芸香苷的标准曲线是用芸香苷的标准溶液,即试剂(2)之9中a、b、c、溶液各1mL,分别加5mL香豆素-甲醇溶液,混合后进行分析。
2.气相色谱仪法
(1)原理:试样先用正己烷脱脂,脱脂后试样用80%甲醇水溶液萃取多酚类。将一部分萃取液浓缩干燥,浓缩物用三甲硅烷基化试剂处理后,进行气相色谱分析。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①正己烷。
②80%甲醇水溶液。
③儿茶酸-甲醇溶液:
a.测定烤烟用:称取1000mg儿茶酸放入200mL的容量瓶中,加80%甲醇水溶液定容至200mL;
b.测定晾晒烟用:将a溶液用80%甲醇溶液稀释10倍即可。
④绿原酸-甲醇溶液:
a.准确称取15mg绿原酸放入10mL的容量瓶中,用80%的甲醇溶液定容到10mL;
b.取5mL溶液a,加③之b溶液2mL;
c.取2mL溶液a,加③之b溶液2mL;
d.取1mL溶液a,加③之b溶液2mL。
⑤三甲基硅烷基化试剂TMS-HT。
(3)方法:
①准确称取约2g的烟叶粉末,用正己烷在索氏萃取器中萃取2h。
②将圆筒滤纸中的脱脂试样用80%甲醇溶液(烤烟为60mL,晾晒烟为30mL)转移到100mL容量的茄型烧瓶中,在沸水浴中回流加热30min。
③向萃取液中加2mL儿茶酸,再用滤纸过滤。
④取一定量的萃取液(烤烟25μL,晾晒烟100μL)放入管式瓶中,在干燥器中用真空泵进行减压浓缩使其干燥,在干燥物中加三甲基硅烷基化试剂TMS-HT(烤烟50μL,晾晒烟30μL),盖紧盖后放入烤箱,在100℃条件下加热1h,使其烷基化,作为气相色谱分析用试样。取试剂b、c、d各40μL,分别加40μL三甲基硅烷基化试剂TMS-HT使其烷基化,作校正曲线用测试样。
⑤气相色谱仪的条件:
气相色谱仪,配有氢火焰离子检测器及数据处理装置。
色谱柱:OV 101,0.4mmi.d.×5m。
温度:200~250℃(5℃/min)。
载气:He,2~3mL/min。
试样注入量:0.5~1μL。
⑥分别用微量注射器吸取校正曲线用溶液注入气相色谱仪进行气相色谱分析。根据峰高比和质量制作校正曲线。
(4)计算:根据校正曲线计算试样中多酚类的含量。
3.比色法
(1)原理:将试样用热水萃取,萃取液用阿尔诺试剂处理后,在510nm下测定吸光度;用氯化铝处理则在416nm下测定吸光度。修正两测定值后,分别计算出绿原酸和云香苷含量。但绿原酸的含量是从多酚的总量中减去云香苷含量来表示的。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①0.5mol/L盐酸溶液。
②阿尔诺试剂:将50g亚硝酸钠和50g钼酸钠(Na2MoO4·2H2O)用500mL水溶解。
③1mol/L氢氧化钠溶液。
④0.1mol/L氯化铝溶液。
⑤绿原酸标准溶液:将100mg绿原酸用30%的甲醇溶解并定容到100mL。分别吸取该溶液1mL、2mL、4mL、6mL,放入50mL容量瓶中,用水定容到50mL。该4种标准溶液每1mL含绿原酸分别为0.02mg、0.04mg、0.08mg、0.12mg。
⑥云香甙标准溶液:将100mg云香甙用30%的甲醇溶解并定容到200mL。分别吸取该溶液2mL、4mL、8mL、12mL,放入50mL容量瓶中,用水定容到50mL。该标准溶液每1mL含云香苷分别为0.02mg、0.04mg、0.08mg、0.12mg。
(3)方法:
①准确称取0.1g烟叶粉末放入试管中,加水约20mL,装上回流冷却装置,在沸水浴中加热萃取1h。
②萃取液冷却后减压过滤,残渣用水洗净后,将洗液和滤液合并放入50mL的烧瓶中,加水定容到50mL。
③准确吸取该溶液4mL放入试管中,共取三组。
④第一试管中加12mL水,作为定量用空白对照。
⑤第二试管中依次加0.5mol/L盐酸溶液4mL,阿尔诺试剂4mL,1mol/L氢氧化钠溶液4mL。
⑥第三试管中加12mL0.1mol/L氯化铝溶液。
⑦准确吸取绿原酸标准溶液及云香苷标准溶液各两个4mL,分别放入试管中,然后按方法⑤及⑥,制备出校正曲线用溶液。
⑧测定吸光度时以水为空白,定量用空白对照在510nm和416nm下测定吸光度。
⑨待测液及标准检测液与阿尔诺试剂反应液在510nm下测定吸光度,而与氯化铝的反应液则在416nm下测定吸光度。
(4)计算:
①将16mL标准检测液中的多酚含量(mg)与吸光度分别作横轴及纵轴。
根据绿原酸标准溶液在510nm和416nm下的吸光度及云香苷标准溶液在510nm和416nm下的吸光度,做4条校正曲线,分别设其斜率为C1、C2、R1、R2。各标准溶液每16mL所含多酚含量分别为0.08mg、0.16mg、0.32mg、0.48mg。
②按下式计算绿原酸的含量w绿原酸,以%表示:

式中 A1——待测液在510nm下的吸光度;
A2——待测液在416nm下的吸光度;
B1——定量用空白在510nm下的吸光度;
B2——定量用空白在416nm下的吸光度;
m——试样的质量,mg。
(十八)烟草中叶绿素的测定
1.原理
将试样用80%的丙酮萃取,利用其对可见光区(663nm、645nm)的吸收进行定量。
2.试剂和材料
所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
(1)80%丙酮(mL/mL)。
(2)碳酸镁或碳酸钙。
3.仪器
经校准的分光光度计。
4.方法
(1)准确称取2~10g试样,与少量碳酸镁或碳酸钙(约0.1g)一起放入40mL的80%丙酮中,用旋转研磨机研磨2min。
(2)将萃取液立即减压过滤,向残渣内加30mL的80%丙酮,再用旋转研磨机研磨2min。
(3)将残渣和萃取液一起进行减压过滤,再用少量80%丙酮洗涤残渣。
(4)将两次滤液放入容量瓶中,再80%丙酮定容到100mL,作为萃取原液。
(5)取一部分萃取原液,用80%丙酮稀释,制成检测液。稀释率应调整到在检测液所使用波长的吸光度为0.2~0.8的倍数范围内为准。
(6)将已制备好的检测液用分光光度计测定在波长663nm及645nm下的吸光度。
5.计算
(1)检测液中叶绿素浓度用下式求出:
将16mL标准检测液中的多酚含量(mg)与吸光度分别作横轴及纵轴。
ρa=12.7A663-2.69A645
ρb=22.9A645-4.68A663
ρ总=8.02A663-20.2A645
式中 ρa、ρb、ρ总——叶绿素a、叶绿素b、总叶绿素的浓度,mg/L;
A663——波长在663nm时的吸光度,光程长1cm;
A645——波长在645nm时的吸光度,光程长1cm。
(2)根据检测液中叶绿素的浓度算出试样中叶绿素的含量。
(十九)烟草中类胡萝卜素的测定
1.薄层层析法
(1)原理:用90%的丙酮萃取色素,用苯转溶后,再用薄层层析分离类胡萝卜素,用分光光度计测定吸光度并定量。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①90%丙酮。
②苯。
③无水硫酸钠。
④食盐。
⑤展开剂:苯∶丙酮=4∶1(体积分数)。
⑥正己烷。
⑦乙醇。
(3)仪器、设备:
①KOH-硅胶薄层板:取20cm×5cm的玻璃板,洗净使其无油脂成分,干燥。将30g硅胶G和70g1%的KOH溶液放入带盖的三角烧瓶中,充分摇混30s。迅速放到薄层板用的涂布器中,在玻璃板上涂0.25mm厚的薄层,风干表面变硬后,即可将板移到干燥箱,在135℃下干燥2h使其获得活性。
②薄层层析展开槽。
③带刻度的小试管。
④分光光度计。
(4)方法:
①准确称取约3g的新鲜烟叶和30mL90%的丙酮一起用旋转研磨机或少量石英砂在研提钵中研磨。
②上部清液用玻璃过滤器减压吸滤,残渣再用丙酮进行研磨,将残渣与萃取液一起用玻璃过滤器减压吸滤。用90%的丙酮冲洗数次直到色素全部被萃取完为止。将萃取液、洗液合并,定容到100mL。
③取一定容积的萃取液(20~25mL)放入分液漏斗中,分液漏斗中预先加好等量的苯。
④将苯和丙酮萃取液混合后,缓缓加水,使其慢慢混合,色素就转移到苯层中去。
⑤去掉水层后,苯层再用水洗涤数次,使混溶的丙酮完全洗掉。如在上下层之间产生乳浊时,可用食盐水代替水。
⑥苯溶液用无水硫酸钠脱水,过滤后放入容量瓶中,定容到50mL。
⑦吸取色素的苯溶液2mL,放入10mL的茄型烧瓶中,用旋转蒸发器在40℃下减压浓缩,最后使其干燥。
⑧向色素中加1~2滴苯使色素溶解,用微量移液管将全部色素加到薄层层析板上。如此反复进行,直到把色素全部转移到薄层板上。该操作应尽快进行。
⑨预先用N2置换展开槽内的空气,放入展开剂,使空间充满展开剂的蒸汽,再将薄层板放入展开槽,约展开15min。
⑩展开完毕后取下薄层板,将吸附有黄色色素的部分分别用小刮勺刮下。
⑾取下的色素放在小号滤纸上,下面接一只带刻度的小试管。β-胡萝卜素用正己烷溶脱,叶黄素、堇菜黄素、新黄素、用乙醇溶脱。然后定容到3~5mL。
⑿上述四个类胡萝卜素组分用分光光度计测定吸光度。个物质的Rf值。最大吸收波长吸光系数见表8-4-3所示:
表8-4-3 各类胡萝卜素的性质

(5)计算:按下式计算各种类被检胡萝卜素的含量ρ胡,以μg/mL表示:

式中 A——各种类胡萝卜素在最大吸收波长的吸光度;
E——各种类胡萝卜素在最大吸收波长的吸光系数(1%浓度时的吸光度)。
2.比色法
(1)原理:用90%的丙酮萃取色素后,用乙醚转溶,用分光光度计测定吸光度并定量。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①90%丙酮。
②乙醚。
③无水硫酸钠。
(3)仪器:分光光度计。
(4)方法:
①准确称取约3g的新鲜烟叶和30mL90%的丙酮一起用旋转研磨机或少量石英砂在研提钵中研磨。
②上部清液用玻璃过滤器减压吸滤,残渣再用丙酮进行研磨,将残渣与萃取液一起用玻璃过滤器减压吸滤。用90%的丙酮冲洗数次直到色素全部被萃取完为止。将萃取液、洗液合并,定容到100mL。
③取一定容积的萃取液(20~50mL)放入分液漏斗中,分液漏斗中预先加好等量的乙醚。
④将乙醚和丙酮萃取液混合后,缓缓加水,使其慢慢混合,色素就转移到乙醚层中去。
⑤去掉水层后,乙醚层再用水洗涤数次,使混溶的丙酮完全洗掉。如在上下层之间产生乳浊时,可用食盐水代替水。
⑥乙醚溶液用无水硫酸钠脱水,过滤后放入容量瓶中,定容到50mL。
⑦稀释浓度要根据被测液在使用波长下,吸光度是否在1以下进行确定。
⑧将稀释好的乙醚溶液用分光光度计测定波长为665nm、649nm、642.5nm、485nm、474nm、470nm时的吸光度。
(5)计算:
被测液中色素浓度用下式求出:

式中 ρ总绿、ρa、ρb、ρ总胡、ρ胡、ρ叶黄——分别为总叶绿素、叶绿素a、叶绿素b、总类胡萝卜素、胡萝卜素、叶黄素的浓度,mg/mL;
A665——波长在665nm时的吸光度,光程长1cm;
A649——波长在649nm时的吸光度,光程长1cm;
A642.5——波长在642.5nm时的吸光度,光程长1cm;
A485——波长在485nm时的吸光度,光程长1cm;
A474——波长在474nm时的吸光度,光程长1cm;
A470——波长在470nm时的吸光度,光程长1cm。
3.高效液相色谱法
(1)原理:试样用70%的甲醇磨碎,再用玻璃过滤器和Sep-pakC18除去不纯物质,吸附的色素用丙酮溶脱,色素用高效液相色谱分离并定量。
(2)试剂:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①70%的甲醇溶液。
②丙酮。
③90%的甲醇溶液(液相色谱用)。
④乙酸乙酯(液相色谱用)。
(3)仪器:
①高效液相色谱仪:
a.柱子: ,3.9mmi.d.×30cm。
,3.9mmi.d.×30cm。
b.移动相:90%的甲醇和乙酸乙酯,将乙酸乙酯在30min内从3%到50%做浓度梯度直线。
c.流速:1.1mL/min。
d.检测器:440nm可见光区的吸光检测器。
e.梯度控制装置。
②5.0μm微孔滤膜。
③Sep-pakC18。
(4)方法:
①准确称取约0.3g的新鲜烟叶和4mL70%甲醇用超声波均化器磨碎均化。
②将磨碎物移入带微孔滤膜的注射器内,注射器前端预先接上Sep-pakC18。
③将甲醇萃取液由注射器中推出,使其通过微孔滤膜及Sep-pakC18。
④残渣再用3mL70%甲醇冲洗2次。
⑤用5mL丙酮溶液解吸出吸附在残渣及Sep-pakC18上的色素。溶出液定容为5mL。
⑥用微量注射器取50μL色素丙酮溶液,注入高效液相色谱仪分离。
⑦定量分析的标准试剂是用薄层层析法分离出的各类胡萝卜素的精制品。先测定各标准试剂的吸光度求出浓度,据此再找出进样量和在色谱图上被分离出的各种类胡萝卜素的峰高(或峰面积)的关系。
(二十)烟草细胞壁构成成分的测定
1.烟草细胞壁物质的制备
(1)原理:从试样中除去水溶性糖类、蛋白质及脂肪类,即留下细胞壁物质。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①乙醇。
②苯。
③丙酮。
④醋酸。
⑤90%二甲亚砜水溶液。
(3)仪器:
①索氏萃取装置。
②搅拌装置。
③离心分离装置。
④玻璃过滤器。
(4)方法:
①乙醇-苯萃取:用筒型滤纸准确称取约5g的干烟叶粉末,放入索氏萃取装置内,加200~250mL乙醇-苯萃取液(1∶2共沸溶液),萃取48h(直到新萃取液不再着色为止)。将萃取残渣放在玻璃过滤器上进行真空过滤,再用乙醇-丙酮冲洗干净,放在五氧化二磷上真空干燥,并称量。
②热水萃取:在上述所得的残渣中加100倍质量的水,装上回流冷却器在沸水浴中加热1h。重复操作3~4次,直到萃取液中没有糖存在为止。将热水萃取后的残渣放在玻璃过滤器上进行真空过滤,过滤后依次用水、乙醇、丙酮清洗,再将残渣放在五氧化二磷上真空干燥,干燥后称量。所得到的物质即为细胞壁物质。
③其他不纯物质的去除:对于含淀粉、蛋白质较多的试样,可按下述鲜烟叶试样的制备进行:
a.用液态氮冻结后再用乳钵粉碎,加50mL1%的脱氧胆酸钠制成悬浊液,然后经过搅拌压榨机将细胞碾碎,再将此悬浊液进行离心分离,残渣用冷的1%的脱氧胆酸钠和冷水洗净。
b.在得到的残渣中加100mL酚-醋酸-水混合液(2∶1∶1),用旋转搅拌机搅拌5min,反复操作两次。
c.在得到的残渣中加100mL90%的二甲亚砜水溶液,用旋转搅拌机搅拌10min,再90%的二甲亚砜水溶液,用旋转搅拌机搅拌10min,再用超声波处理。混合物在室温条件下搅拌16h后离心分离,其残渣用90%的二甲亚砜水溶液、水洗净,再用乙醇-丙酮冲洗,干燥后称量,所得到的物质即为细胞壁物质。
2.果胶质的定量
(1)原理:细胞壁构成成分中能用乙二胺四乙酸钠(EDTA-Na2)萃取的成分即为果胶质。果胶质的量可用两种方法确定:
①用比色法求出萃取液中半乳糖醛酸的含量,该量即为果胶质的含量;
②将萃取液透析脱盐后再经冷冻干燥,求其质量,即为果胶质含量。
(2)试剂和材料:
所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①0.05mol/L乙二胺四乙酸钠(EDTA-Na2)溶液:在0.05mol/L醋酸钠缓冲溶液中(pH=4.5)溶解乙二胺四乙酸钠,使其浓度达到0.05mol/L。
②硫酸溶液:将0.04g的特级硼酸钠(NaB4O7·10H2O)溶解在100mL特级硫酸中,其浓度为0.0125mol/L。
③0.15%的对羟基联苯:将0.15g的对羟基联苯用100mL0.5%的氢氧化钠溶液溶解于暗处可保存1个月。
(3)仪器:分光光度计。
(4)方法:
①准确称取2g细胞壁物质,加EDTA-Na2溶液200mL,装上回流冷却装置在沸水浴上加热1h。重复操作3~4次,直到萃取液中没有糖存在为止。冷却后将反应液用玻璃过滤器真空过滤,残渣用水洗净,将滤液和洗液收集后定容。
②取6mL硫酸溶液放在试验管中,用冰充分冷却后,将1mL滤液小心地加在硫酸溶液上面。在低于室温条件下使其充分冷却,同时轻轻振动,然后再逐渐加快振动使其混合。
③取下玻璃球型盖后在沸水浴上加热5min。再在冰水中冷却,然后加0.1mL对羟基联苯,搅拌后呈红色。1h后在520nm测定吸光度。该颜色可稳定12h。
④将方法①得到的溶液浓缩到50~100mL后,先用自来水,然后用蒸馏水透析,冷冻干燥后求质量。
3.全纤维素的定量
(1)原理:将果胶萃取残渣用冰醋酸和次氯酸钠进行脱木质素处理,其残渣即为全纤维素。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①冰醋酸。
②次氯酸钠。
③丙酮。
④0.05mol/L乙二胺四乙酸钠(EDTA-Na2)溶液:在0.05mol/L醋酸钠缓冲溶液中(pH=4.5)溶解乙二胺四乙酸钠,使其浓度达到0.05mol/L。
(3)方法:
①准确称取2g细胞壁物质,加乙二胺四乙酸钠溶液200mL,装上回流冷却装置在沸水浴上加热1h。重复操作3~4次,直到萃取液中没有糖存在为止。冷却后将反应液用玻璃过滤器真空过滤,残渣用水洗净。
②将残渣放入300mL的烧瓶中,加蒸馏水100mL,冰醋酸0.2mL,次氯酸钠0.06g后,松松地盖上瓶栓,放在70~80℃的水浴上加热1h,加热过程中要经常轻轻振动。
③白色残渣用预先称量的玻璃过滤器过滤。再用水及丙酮洗净,干燥后称量。
4.半纤维素的定量
(1)原理:将果胶萃取残渣用冰醋酸和次氯酸钠进行脱木质素处理,其残渣即为全纤维素。将全纤维素用氢氧化钾萃取,可溶性部分即为半纤维素。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①冰醋酸。
②次氯酸钠。
③丙酮。
④0.05mol/L乙二胺四乙酸钠(EDTA-Na2)溶液:在0.05mol/L醋酸钠缓冲溶液中(pH=4.5)溶解乙二胺四乙酸钠,使其浓度达到0.05mol/L。
⑤4%氢氧化钾溶液。
⑥44%氢氧化钾溶液。
⑦稀醋酸溶液。
⑧苯酚水溶液:将5g特级苯酚用蒸馏水溶解,并稀释到100mL。
⑨浓硫酸。
(3)方法:
①准确称取2g细胞壁物质,加乙二胺四乙酸钠溶液200mL,装上回流冷却装置在沸水浴上加热1h。重复操作3~4次,直到萃取液中没有糖存在为止。冷却后将反应液用玻璃过滤器真空过滤,残渣用水洗净。
②将残渣放入300mL的烧瓶中,加蒸馏水100mL,冰醋酸0.2mL,次氯酸钠0.06g后,松松地盖上瓶栓,放在70~80℃的水浴上加热1h,加热过程中要经常轻轻振动。
③白色残渣用预先称量的玻璃过滤器过滤。再用水及丙酮洗净,干燥后称量。
④在残渣中加入50mL4%氢氧化钾溶液,在室温下通氮气搅拌。
⑤4h后再加入50mL44%氢氧化钾溶液再在室温下通氮气搅拌一夜。
⑥残渣用预先称量的玻璃过滤器过滤,并用水、醋酸溶液洗净,将滤液和洗液合并。
⑦取1mL试样溶液(调整成含有10~100μg的中性糖)放入试管中,加入1mL苯酚溶液并混合,再向溶液中加5mL的浓硫酸,加入时不要使硫酸接触管壁,而应直接、迅速地加到液面上,充分混合后在室温条件下放置30min,再在490nm处测定吸光度,以葡萄糖量表示。
⑧将方法⑥得到的溶液用醋酸中和(pH5~6)后,浓缩至50~100mL,先用自来水,然后用蒸馏水透析,冷冻干燥后求质量。
5.α-纤维素的定量
(1)原理:将果胶萃取残渣用冰醋酸和次氯酸钠进行脱木质素处理,其残渣即为全纤维素。将全纤维素用氢氧化钾萃取,残渣部分即为α-纤维素。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①冰醋酸。
②次氯酸钠。
③丙酮。
④0.05mol/L乙二胺四乙酸钠(EDTA-Na2)溶液:在0.05mol/L醋酸钠缓冲溶液中(pH=4.5)溶解乙二胺四乙酸钠,使其浓度达到0.05mol/L。
⑤4%氢氧化钾溶液。
⑥44%氢氧化钾溶液。
⑦稀醋酸溶液。
⑧苯酚水溶液:将5g特级苯酚用蒸馏水溶解,并稀释到100mL。
⑨浓硫酸。
(3)方法:
①准确称取2g细胞壁物质,加乙二胺四乙酸钠溶液200mL,装上回流冷却装置在沸水浴上加热1h。重复操作3~4次,直到萃取液中没有糖存在为止。冷却后将反应液用玻璃过滤器真空过滤,残渣用水洗净。
②将残渣放入300mL的烧瓶中,加蒸馏水100mL,冰醋酸0.2mL,次氯酸钠0.06g后,松松地盖上瓶栓,放在70~80℃的水浴上加热1h,加热过程中要经常轻轻振动。
③白色残渣用预先称量的玻璃过滤器过滤。再用水及丙酮洗净,干燥后称量。
④在残渣中加入50mL4%氢氧化钾溶液,在室温下通氮气搅拌。
⑤4h后再加入50mL44%氢氧化钾溶液再在室温下通氮气搅拌一夜。
⑥残渣用预先称量的玻璃过滤器过滤,并用乙醇、丙酮冲洗,干燥后称量。
6.木质素的定量
(1)原理:细胞壁物质的硫酸水解残渣即为木质素。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
72%硫酸。
(3)方法:
①准确称取0.5g细胞壁物质放入200mL的烧杯中,加10mL72%的硫酸,充分搅拌,在20℃条件下放置4h。
②将上述试样定量移入500mL的茄型烧瓶中,加水后使硫酸稀释到3%,装上回流冷却装置煮沸2h。
③水解残渣放冷后,放入已称量的玻璃过滤器中过滤,然后清洗残渣,干燥后称量即为粗木质素质量。
(二十一)烟草粗灰分的测定
1.原理
将试样在550℃下灰化,残渣即作为粗灰分并进行定量。
2.仪器
使用马福炉。
3.方法
①准确称取约2g试样,放入预先经550℃高温恒量的坩埚内。
②将坩埚放入马福炉慢慢升温至350℃,稍停一段时间,使试样初步灰化(产生烟雾),然后升温至550℃,灰化2h。
③放入干燥器中冷却后称量。
4.计算
粗灰分用下式算出:

式中 H——粗灰分,%;
m′——灰分质量,g;
m——试样质量,g。
(二十二)无机成分分析用试样的制备
1.干式灰化法(Ⅰ)
(1)分析对象:钾、钙、镁、铁、锰、铝、硼、铜、钠。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
盐酸。
(3)方法:
①用坩埚准确称取1g的试样,盖上盖放入马福炉中,慢慢加温至350℃,稍停一段时间,使试样初步灰化,然后升温至450℃左右,灰化2h。
②冷却后用水将灰分冲入100mL的烧杯中,用5mL盐酸冲洗坩埚,洗液也放入烧杯中,盖上表面皿,煮沸数分钟。
③用滤纸过滤,烧杯及滤纸上的残渣用热水反复冲洗,将滤液和洗液合并后定容到100~200mL,即得到灰化萃取原液。
2.干式灰化法(Ⅱ)
(1)分析对象:磷。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①硝酸镁溶液:将95g硝酸镁用水溶解,定容到100mL。
②硝酸。
③盐酸。
(3)方法:
①用坩埚准确称取约1g的试样,加硝酸镁溶液1mL。
②放在沸水浴上数分钟后,小心加数滴盐酸,直到使其炭化。在沸水浴上溶液发粘后,放在沙浴上使其完全干固。
③盖上坩埚盖,放入马福炉中,将温度升到500℃灼烧直到得到灰色的灰。如有必要可在坩埚冷却后加少量水或乙醇与甘油混合液,使灰分溶解并干固。然后不盖盖放入马福炉中加热。
④冷却后用少量水将灰分移入100mL的烧杯中,加10mL硝酸,盖上表面皿,煮沸数分钟。
⑤用滤纸过滤,烧杯及滤纸上的残渣用热水反复冲洗,将滤液和洗液合并后定容到100~200mL,即得到灰化萃取原液。
3.湿式灰化法
(1)分析对象:磷、钾、钙、镁、铁、锰、铝、硼、铜、钠。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①硝酸。
②高氯酸。
(3)方法:
①准确称取1g的试样放入长颈烧瓶中,加10mL硝酸,温火加热分解30min后冷却。
②冷却后加5mL高氯酸,在继续温火加热分解,当溶液达到无色或接近无色,出现白烟即可认为分解完全,停止加热将其冷却。
③用纯水将长颈烧瓶中的溶液移入100~200mL的容量瓶中,并定容,即得到灰化原液。如果不溶物多时,需过滤后才能供分析用。
4.萃取法
(1)分析对象:钾、钠。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①醋酸镁-醋酸铵萃取用原液:将308g醋酸铵和42.8g醋酸镁用水溶解,在用醋酸或氨水调整pH在6.7~7.1后,定容到1L。
②萃取溶液:将萃取原液用水稀释2倍。
(3)方法:用200mL三角烧瓶准确称取1g的试样,准确加100mL萃取原液,振动萃取2h,用滤纸过滤,即得到萃取原液。
(二十三)烟草中钾的测定
1.火焰光度计法
(1)原理:将试样灰化萃取液或醋酸镁-醋酸铵萃取液进行火焰光度分析。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
钾的标准溶液:将氯化钾在105℃下干燥数小时后,准确称取1.907g,用水溶解后定容到1L。该溶液含钾1000×10-6mg/kg。用该溶液制成0、2×10-6mg/kg、4×10-6mg/kg、6×10-6mg/kg、8×10-6mg/kg、10×10-6mg/kg的钾的标准溶液。被测液无论是用灰化萃取液还是用醋酸镁-醋酸铵萃取液制备,都必须往钾的标准溶液添加与被测液相同的酸或盐类。
(3)仪器:火焰光度计。
(4)方法:
①用无机成分分析用试样的制备干式灰化法(Ⅰ)或湿式灰化法制备的灰化萃取原液配制测定用原液时,钾的浓度应在标准溶液的浓度范围内,必要时可用水稀释。
②在测定条件下,先用火焰光度计测定标准溶液,然后再测定被测液。
③根据钾的标准溶液的读数做出校正曲线,再从被测液的读数求出被测液中钾的浓度。
(5)计算:按下式计算烟草中钾的质量分数wk,以%表示:

式中 cp——被测液中钾的浓度,10-6mg/kg;
V——灰化或萃取原液的体积,mL;
d——稀释率;
m——试样质量,g。
2.原子吸收光谱法
(1)原理:将试样灰化萃取液用原子吸收光谱仪分析。
(2)仪器:原子吸收光谱仪,分析线为766.5nm的光源。
(3)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
钾的标准溶液:将氯化钾在105℃下干燥数小时后,准确称取1.907g,用水溶解后定容到1L。该溶液含钾1000×10-6mg/kg。用该溶液制成0、2×10-6mg/kg、4×10-6mg/kg、6×10-6mg/kg、8×10-6mg/kg、10×10-6mg/kg的钾的标准溶液。被测液无论是用灰化萃取液还是用醋酸镁-醋酸铵萃取液制备,都必须往钾的标准溶液添加与被测液相同的酸或盐类。
(4)方法:
①用无机成分分析用试样的制备干式灰化法(Ⅰ)或湿式灰化法制备的灰化萃取原液配制测定用原液时,钾的浓度应在标准溶液的浓度范围内,必要时可用水稀释。
②将钾的标准溶液和被测液用原子吸收光谱仪分析。
③根据钾的标准溶液的读数做出校正曲线,再从被测液的读数求出被测液中钾的浓度。
(5)计算:按下式计算烟草中钾的百分含量wK,以%表示:

式中 cp——被测液中钾的浓度,10-6mg/kg;
V——灰化或萃取原液的体积,mL;
d——稀释率;
m——试样质量,g。
(二十四)烟草中钠的测定——火焰光度计法
1.原理
将试样灰化萃取液或醋酸镁-醋酸铵萃取液进行火焰光度分析。
2.试剂和材料
所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
钠的标准溶液:将氯化钠在105℃下干燥数小时后,准确称取2.542g,用水溶解后定容到1L。该溶液含钠1000×10-6mg/kg。用该溶液制成0、20×10-6mg/kg、40×10-6mg/kg、60×10-6mg/kg、80×10-6mg/kg、100×10-6mg/kg的钠的标准溶液。被测液无论是用灰化萃取液还是用醋酸镁-醋酸铵萃取液制备,都必须往钠的标准溶液添加与被测液相同的酸或盐类。
3.仪器
火焰光度计。
4.方法
(1)用无机成分分析用试样的制备干式灰化法(Ⅰ)或湿式灰化法制备的灰化萃取原液配制测定用原液时,钠的浓度应在标准溶液的浓度范围内,必要时可用水稀释。
(2)在测定条件下,先用火焰光度计测定标准溶液,然后再测定被测液。
(3)根据钠的标准溶液的读数做出校正曲线,再从被测液的读数求出被测液中钠的浓度。
5.计算
按下式计算烟草中钠的质量分数wNa,以%表示:
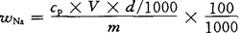
式中 cp——被测液中钠的浓度,10-6mg/kg;
V——灰化或萃取原液的体积,mL;
d——稀释率;
m——试样质量,g。
(二十五)烟草中钙的测定
1.原子吸收光谱法
(1)原理:将试样灰化萃取液用原子吸收光谱法分析。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①氯化镧溶液:将134g氯化镧(LaCl3·7H2O)用水溶解后定容到1L。该溶液含镧50000×10-6mg/kg。取2mL该溶液加到被测液中并定容到100mL。被测液中含镧1000×10-6mg/kg。
②钙的标准溶液:称取2.497g碳酸钙,加水约50mL后,缓缓加入5mL盐酸(1+1)使之溶解,用水定容到1L。该溶液含钙1000×10-6mg/kg。用该溶液制成0、2×10-6mg/kg、4×10-6mg/kg、6×10-6mg/kg、8×10-6mg/kg、10×10-6mg/kg钙的系列标准溶液。同时分别加入2mL氯化镧溶液,使各标准溶液都含1000×10-6mg/kg的镧。
(3)仪器:原子吸收光谱仪,分析线为422.7nm的光源。
(4)方法:
①用容量瓶称取一定量的无机成分分析用试样制备的干式灰化法(Ⅰ)或湿式灰化法制备的灰化萃取原液。称取的量应使被测液中钙的浓度在标准溶液的系列浓度范围内,再适当添加氯化镧溶液,调整被测液中镧的含量,使其仍含1000×10-6mg/kg。然后再定容。
②将钙的标准溶液和被测液用原子吸收光谱法分析。
③根据钙的标准溶液的各浓度及其读数做出校正曲线,再从被测液的读数求出被测液中钙的浓度。
(5)计算:
按下式计算烟草中钙的质量分数wca,以%表示:

式中 cp——被测液中钙的浓度,10-6mg/kg;
V——灰化或萃取原液的体积,mL;
d——稀释率;
m——试样质量,g。
2.螯合滴定法
(1)原理:将试样灰化萃取液的pH调整到10,用乙二胺四乙酸溶液滴定,求出钙与镁的总量。另外再取一份萃取液加入能使钙沉淀的试剂,过滤后除去钙,余下的溶液仍用乙二胺四乙酸溶液滴定,求出镁的含量,上述二物质的总量与镁含量的差即为钙的含量。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①氨水(1+2)。
②醋酸溶液(1+9)。
③pH10缓冲液:将70g氯化胺和570mL浓氨水用水溶解后定容到1L。
④钙沉淀剂:将140g氯化胺和6g草酸胺用水溶解后定容到1L。
⑤2%的二氯氧化锆溶液:将2g的二氯氧化锆溶解在100mL水中。
⑥三乙醇胺溶液(1+1)。
⑦0.01mol/L钙的标准溶液:将特级碳酸钙放入烘箱,以110℃干燥,准确称取1.0000g碳酸钙,加少量盐酸,加温使其溶解,定容到1L。
⑧0.01mol/L乙二胺四乙酸溶液:将3.723g乙二胺四乙酸钠用水溶解后定容到1L。该溶液用钙的标准溶液标定,求出系数。
⑨甲基红指示剂:将0.1g甲基红溶解在100mL乙醇中。
⑩BT指示剂:将0.5g羊毛铬黑T色素溶解在500mL三乙醇胺中,放入褐色瓶中保存。
(3)方法:
①从无机成分分析用试样制备的干式灰化法(Ⅰ)或湿式灰化法制备的灰化萃取原液中取一定量的原液(应含镁0.5~2mg)加数滴甲基红指示剂,再用氨水调整成碱性。
②加醋酸溶液呈微酸性后,将溶液加温,同时根据钙的含量加10~30mL钙沉淀剂,然后放置4h。
③将生成的草酸钙沉淀用滤纸过滤,充分洗净后,将滤液和洗液合并,放入100mL的三角烧瓶中。
④将除去钙的滤液用磁性搅拌器搅拌,并加入5mL缓冲液和2mL三乙醇胺溶液,混合后再加2滴BT指示剂。
⑤立刻用0.01mol/L乙二胺四乙酸溶液滴定,接近终点时溶液由红紫色逐渐变成蓝色,红色完全消失即为终点。接近终点时滴定要慢一些。该滴定值为镁的滴定数。设为AmL。
⑥再取一定量的萃取原液(80mL以下)放入100mL容量瓶中,加数滴甲基红指示剂,再加2mL二氯氧化锆溶液,用氨水调整pH为6(红色消失),再用水定容。
⑦将上述溶液用滤纸过滤到三角烧瓶中,沉淀物不用冲洗,舍去不要。
⑧吸取一定量的滤液,按方法④、⑤的方法进行滴定,设为BmL。
(4)计算
按下式计算烟草中钙和镁的质量分数w镁、w钙,以%表示:

1mL0.01mol/L乙二胺四乙酸溶液=0.4008mgCa
1mL0.01mol/L乙二胺四乙酸溶液=0.2432mgMg
式中 A——镁的滴定数,mL;
B——钙、镁总量的滴定数,mL;
V1——方法(3)之⑥中吸取的液体量,mL;
V2——方法(3)之⑧中吸取的液体量,mL;
Vt——供滴定的被测液量,mL;
V——灰化萃取原液的量,mL;
f——0.01mol/L乙二胺四乙酸溶液系数;
m——试样质量,g。
(二十六)烟草中镁的测定
1.原子吸收光谱法
(1)原理:将试样灰化萃取液用原子吸收光谱法分析。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①氯化镧溶液:将134g氯化镧(LaCl3·7H2O)用水溶解后定容到1L。该溶液含镧50000×10-6mg/kg。取2mL该溶液加到被测液中并定容到100mL。被测液中含镧1000×10-6mg/kg。
②镁的标准溶液:准确称取1.000g特级金属镁,溶解在稀盐酸中,用水定容到1L。该溶液含镁1000×10-6mg/kg。用该溶液制成0、0.2×10-6mg/kg、0.4×10-6mg/kg、0.6×10-6mg/kg、0.8×10-6mg/kg、1.0×10-6mg/kg镁的系列标准溶液。再分别加入2mL氯化镧溶液,使各标准溶液都含1000×10-6mg/kg的镧。
(3)仪器:原子吸收光谱仪,分析线为285.2nm的光源。
(4)方法:
①用容量瓶称取一定量的无机成分分析用试样制备的干式灰化法(Ⅰ)或湿式灰化法制备的灰化萃取原液。称取的量应使被测液中镁的浓度在标准溶液的系列浓度范围内,再适当添加氯化镧溶液,调整被测液中镧的含量,使其仍含1000×10-6mg/kg。然后再定容。
②将镁的标准溶液和被测液用原子吸收光谱法分析。
③根据镁的标准溶液的各浓度及其读数做出校正曲线,再从被测液的读数求出被测液中镁的浓度。
(5)计算:按下式计算烟草中镁的质量分数w镁,以%表示:

式中 cp——被测液中镁的浓度,10-6mg/kg;
V——灰化或萃取原液的容量,mL;
d——稀释率;
m——试样质量,g。
2.螯合滴定法
与烟草中钠的测定相同。
(二十七)烟草中磷的测定
1.原理
用钒钼酸铵混合试剂使磷酸变色,进行变色定量。
2.试剂和材料
所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
钒钼酸铵硝酸溶液:
(1)将25.0g钼酸铵溶于大约400mL温水中。
(2)将1.25g偏钒酸铵放入约300mL水中,煮沸使其溶解,冷却后加入250mL硝酸。
(3)将(1)液倒入(2)液中,加水定容到1L。
磷酸标准溶液:将磷酸二氢钾放入烘箱,在105℃烘2h,准确称取0.2195g放入1L的容量瓶中,加水约400mL使其溶解后,再加25mL硫酸,定容到1L。
3.方法
(1)从磷酸标准溶液中分别取2mL、4mL、6mL、8mL、10mL(分别含磷0.1mg、0.2mg、0.3mg、0.4mg、0.5mg)放入50mL的容量瓶中,各加10mL钒钼酸铵硝酸溶液使其显色,加水定容到50mL。
(2)取5~10mL用干式灰化法(Ⅰ)或湿式灰化法制备的灰化萃取原液放入容量瓶中,加10mL钒钼酸铵硝酸溶液使其显色,加水定容到50mL。
(3)显色后的溶液至少放置10min,再用420~440nm波长进行比色。做出校正曲线,再从被测液的吸光度求出试样中磷的含量。
比色时的空白对照只加钒钼酸铵硝酸溶液。
4.计算
按下式计算烟草中磷的质量分数w磷,以%表示:

式中 mp——被测液中磷的含量,mg;
V——灰化或萃取原液的体积,mL;
V′——供显色被测液量,mL;
m——试样质量,g。
(二十八)烟草中氯含量的测定——佛尔哈德法
1.原理
烟草试样经浓硝酸硝化分解,氯离子与预先定量加入的硝酸银标准溶液生成氯化银沉淀,以三价铁离子为指示剂,用硫氰酸铵标准溶液滴定剩余的硝酸银,从而求出试样中氯的含量。反应式如下:
Ag++C1-=AgCl↓
Ag++CNS-=AgCNS↓
过量的硫氰酸铵与三价铁离子成生微棕色络合物,即为终点:
3CNS-+Fe3+=Fe(CNS)3
2.试剂和材料
所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
(1)浓硝酸。
(2)高锰酸钾饱和溶液。
(3)铁铵矾[NH4Fe(SO4)22H2O]饱和溶液。
(4)硫氰酸铵标准溶液。
(5)硝酸银标准溶液。
(6)1%氯化钠溶液。
3.仪器
(1)回流冷凝装置。
(2)10mL和15mL单刻度移液流管。
4.方法
(1)硫氰酸铵标准溶液的制备:
①配制:称取7.7g硫氰酸铵溶于1000mL水中,摇匀。
②标定:称取0.5g于硫酸干燥器中干燥到恒量的基准硝酸银,称准至0.0001g,溶于100mL水中,加2mL硫酸铁铵指示液(80g/L)及10mL硝酸溶液(25%),在摇动下用配制好的硫氰酸铵溶液[c(NH4CNS)=0.1mol/L]滴定,终点前摇动溶液至完全清亮后,继续滴定至溶液所呈浅棕红色保持30s。
③计算:按下式计算硫氰酸铵标准溶液浓度:

式中 c(NH4CNS)——硫氰酸铵标准溶液浓度,mo1/L;
m——硝酸银质量,g;
V——硫氰酸铵溶液用量,mL;
0.1699——与1.00mL硫氰酸铵标准溶液[c(NH4CNS)=1.000mol/L]相当的以g表示的硝酸银质量。
浓度值应精确至0.0001mol/L。
平行实验不得少于8次,两人各做4次,每人4次平行测定结果的极差与平均值之比不得大于0.1%,两人测定结果平均值之差不得大于0.1%。结果取平均值。
(2)硝酸银标准溶液的制备:
①配制:称取17.5g硝酸银,溶于1000mL水中,摇匀,溶液保存于棕色瓶中。
②标定:量取30.00~35.00mL,的硝酸银溶液,加70mL水,1mL硫酸铁铵指示液(80g/L)及10mL硝酸溶液(25%),在摇动下用硫氰酸铵标准溶液滴定,终点前摇动溶液至完全清亮后,继续滴定至溶液所呈浅棕色保持30s。
(3)计算:按下式计算硝酸银标准溶液浓度:

式中 c(AgNO3)——硝酸银标准溶液浓度,mol/L;
V1——硫氰酸铵标准溶液的用量,mL;
c1——硫氰酸铵标准溶液浓度,mol/L;
V——硝酸银标准溶液的用量,mL。
浓度值应精确至0.0001mol/L。
平行实验不得少于8次,两人各做4次,每人4次平行测定结果的极差与平均值之比不得大于0.1%,两人测定结果平均值之差不得大于0.1%。结果取平均值。
(4)测定:称取烟草试样1~2g于250mL三角瓶内,称准至0.001g用单刻度移液管移取10mL硝酸银标准溶液于三角瓶中(试样多或氯含量高时可移取15mL),加入浓硝酸30mL,饱和高锰酸钾溶液5mL。在通风橱内,将三角瓶置于电炉上,连接回流冷凝装置,加热硝化30min。然后,用水冲洗冷凝管内壁及末端,取下三角瓶,将硝化液过滤于烧杯中,用水充分洗涤沉淀,用氯化钠溶液检查硝酸银已被完全洗脱。向烧杯中加入5mL铁铵矾溶液,用硫氰酸铵标准溶液滴定至溶液呈微棕色。
5.计算
按下式计算烟草中氯的质量分数:

式中 wC1——烟草中氯的质量分数,%;
c1——硝酸银标准溶液浓度,mol/L;
c2——硫氰酸铵标准溶液浓度,mol/L;
V1——加入三角瓶中的硝酸银标准溶液体积,mL;
V2——硫氰酸铵标准溶液用量,mL;
m——试样质量,g;
w——试样的含水率,%;
35.5——氯的摩尔质量,g/mol。
结果以平行测定的平均值报出,精确至0.01%。
(二十九)烟草中硫的测定
1.原理
将试样与硝酸镁混合后灰化,再从其萃取液中将硫以硫酸钡的形式沉淀下来,然后用质量法进行定量。
2.试剂和材料
所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
(1)硝酸镁溶液:取95g硝酸镁溶于水,定容到100mL。
(2)盐酸。
(3)10%的氯化钡溶液:将11.6g的氯化钡溶于水,定容到100mL。
3.方法
(1)在100mL镍坩埚中准确称取约3g试样,加入硝酸镁溶液,其加入量仅限于将试样充分湿润即可。
(2)在105℃下使水分蒸发,然后放在沙浴上加热。如果试样出现膨胀应立即从沙浴上拿下来,然后再放上去加热,直到有褐色的氧化氮气体逸出为止。
(3)将马福炉的温度控制在500℃以下,放入试样加热,使其全部氧化,直到成为黑色粒状物为止。
(4)冷却后加水,再加入约5mL的盐酸煮沸。
(5)用滤纸过滤,充分洗净残留物。
(6)将滤液和洗液合并,用水定容到200mL。
(7)将滤液加热,一边搅拌一边加入10%的氯化钡溶液10mL。
(8)煮沸5min后,在沸水浴下放置5h或长时间地放在暖处,以待沉淀。
(9)用滤纸或古氏坩埚过滤。
(10)沉淀用热水冲洗,直到与氯离子不反应为止。
(11)将滤纸与沉淀一起烘干,再放入坩埚在马福炉中灰化,冷却后称量。该量为硫酸钡的质量。
4.计算
按下式计算烟草中硫的质量分数w硫,以%表示:

式中 m′——硫酸钡的质量,g;
m——试样质量,g。
(三十)烟草中铁的测定
1.原子吸收光谱法
(1)原理:将试样灰化萃取液用原子吸收光谱法分析。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
铁的标准溶液:准确称取1g纯铁,放入约30mL2mol/L盐酸中煮沸溶解后,再放入1L的容量瓶中,用水溶解后定容到1L。该溶液含铁1000×10-6mg/kg。用该溶液制成0、2×10-6mg/kg、4×10-6mg/kg、6×10-6mg/kg、8×10-6mg/kg、10×10-6mg/kg的铁的标准溶液。
(3)仪器:原子吸收光谱仪,分析线为248.3nm的光源。
(4)方法:
①用无机成分分析用试样的制备干式灰化法(Ⅰ)或湿式灰化法制备的灰化萃取原液直接进行测定。铁含量高时应将其稀释到铁标准溶液的浓度范围内。
②将铁的标准溶液和待检液用原子吸收光谱法分析。
③根据铁的标准溶液的各浓度及其读数做出校正曲线,再从被测液的读数求出被测液中铁的浓度。
(5)计算
按下式计算烟草中铁的含量ρ铁,以10-6mg/kg表示:

式中 cp——被测液中铁的浓度,10-6mg/kg;
V——灰化或萃取原液的容量,mL;
d——稀释率;
m——试样质量,g。
2.比色法
(1)原理:将试样灰化萃取液中的铁用对苯二酚还原,以二氮杂菲使其显色,并进行比色定量。
(2)试剂和材料:所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
①2mol/L醋酸溶液。
②2mol/L醋酸钠溶液。
③1%柠檬酸二胺溶液。
④pH3.5醋酸缓冲溶液:将6.4mL2mol/L醋酸钠溶液和93.6mL2mol/L醋酸溶液混合,加水到1L。
⑤pH4.5醋酸缓冲溶液:将43mL2mol/L醋酸钠溶液和57mL2mol/L醋酸溶液混合,加水到1L。
⑥对苯二酚溶液:将1g对苯二酚用pH4.5醋酸缓冲溶液溶解,放冰箱中保存。如有显色现象应扔掉重新配制。
⑦二氮杂菲溶液:将1g二氮杂菲溶解并定容400mL。溶解时可根据需要加热。
⑧铁的标准溶液:准确称取0.1g纯铁,用约30mL的2mol/L盐酸煮沸溶解,放入容量瓶中用水定容到1L。该溶液每1mL含铁0.1mg。
⑨溴酚蓝指示剂:将0.1g溴酚蓝(B.P.B.)和3mL0.05mol/L氢氧化钠放入乳体中研磨混合,用水溶解后定容到250mL。
(3)仪器:分光光度计。
(4)方法:
①准确称取0.1g纯铁用无机成分分析用试样的制备干式灰化法(Ⅰ)或湿式灰化法制备的灰化萃取原液2份,分别放入25mL容量瓶及100mL三角瓶中。萃取原液的量应保证铁的含量在0.02~0.1mg。
②向三角烧瓶的溶液中加数滴溴酚蓝指示剂,再滴加2mol/L醋酸溶液,使其pH为 3.5左右,并求出所需醋酸钠的溶液量。
③向容量瓶的溶液中加1mL对苯二酚溶液和2mL二氮杂菲溶液,再加入与(4)之②等量的2mol/L醋酸溶液,使其pH为3.5左右。
④如果在三角烧瓶中调节pH后,溶液出现混浊的话,需在加醋酸钠溶液之前,加1%柠檬酸二胺溶液1mL。
⑤加水定容到25mL,放置1h。
⑥用分光光度计在波长为510nm下测定吸光度,或用绿色滤片进行比色定量。
铁的标准溶液也按上述方法操作,分别对含0~0.1mg铁的溶液显色测定后,做成校正曲线再根据被测液的吸光度求出铁的含量。
(5)计算:
按下式计算烟草中铁的含量ρ铁,以10-6mg/kg表示:

式中 mp——被测液中铁的含量,mg;
V——灰化萃取原液的量,mL;
Vt——供显色用的被测液量,mL;
m——试样质量,g。
(三十一)烟草中铝的测定
1.原理
用试铝灵将样中的铝制螯合物进行比色定量。
2.试剂和材料
所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
(1)试铝灵溶液:
①将0.5g试铝灵用水溶解。
②取10g阿拉伯胶用200mL水溶解后过滤。
③将100g醋酸铵溶解于水并定容为400mL再用醋酸或氨水调整pH为4.5。
(2)消泡剂溶液:称取0.03g硅消泡剂溶解在100mL水中。
(3)1%巯基乙酸水溶液。
(4)铝的标准液:准确称取0.1g金属铝,放入100mL的烧杯中,加6mL盐酸,盖上表面皿慢慢加热,使铝完全溶解。冷却后加水到1L。每1mL该溶液含铝100μg。使用时再稀释10倍,即使每1mL该溶液含铝10μg。
3.仪器
分光光度计。
4.方法
(1)取一定量用无机成分分析用试样的制备干式灰化法(Ⅰ)或湿式灰化法制成的灰化萃取原液,放入100mL的容量瓶中,加水20mL稀释。
(2)加1%巯基乙酸2mL、消泡剂溶液0.5mL、试铝灵溶液10mL。
(3)在沸水上加热20min。
(4)冷却30min后加水到100mL。
(5)用分光光度计测定在525nm处的吸光度。
(6)用铝的标准溶液配制含0μg、20μg、40μg、60μg、80μg、100μg铝的标准检测样,并显色测定,制成校正曲线。再根据试样被测液的吸光度求出试样中的铝含量。
5.计算
按下式计算烟草中铝的含量ρ铝,以10-6mg/kg表示:

式中 mp——被测液含铝量,μg;
V。——灰化萃取原液的体积,mL;
Vt——供显色用被测液量,mL;
m——试样质量,g。
(三十二)烟草中锰的测定——原子吸收光谱法
1.原理
将试样灰化萃取液用原子吸收光谱法分析。
2.试剂和材料
所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
锰的标准溶液:准确称取1g纯锰,取少量稀盐酸溶解,再用水稀释到1L。该溶液含锰1000×10-6mg/kg。用该溶液制成0、2×10-6mg/kg、4×10-6mg/kg、6×10-6mg/kg、8×10-6mg/kg、10×10-6mg/kg的铁的标准溶液。
3.仪器
原子吸收光谱仪,分析线为279.5nm的光源。
4.方法
(1)取一定量的用无机成分分析用试样的制备干式灰化法(Ⅰ)或湿式灰化法制备的灰化萃取原液,稀释成含锰量为2×10-6mg/kg~10×10-6mg/kg的溶液。
(2)将锰的标准溶液和待检液用原子吸收光谱法分析。
(3)根据锰的标准溶液的各浓度及其读数做出校正曲线,再从被测液的读数求出被测液中锰的浓度。
5.计算
按下式计算烟草中锰的含量ρ锰,以10-6mg/kg表示:
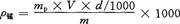
式中 mp——被测液中锰的浓度,10-6mg/kg;
V——灰化或萃取原液的容量,mL;
d——稀释率;
m——试样质量,g。
(三十三)烟草中硼的测定
1.原理
利用硼与姜黄素在酸性条件下加热蒸发时产生的红色色素进行比色定量。
2.试剂和材料
所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
(1)0.1mol/L盐酸。
(2)0.1mol/L氢氧化钙悬浊液:称取1.85g氢氧化钙加水500mL,充分摇混。
(3)盐酸-草酸混合液:取50g草酸加水200mL摇混,放置一日。取其上部清液80mL与20mL盐酸混合。
(4)0.1%姜黄素溶液:称取0.1g姜黄素,用100mL95%乙醇溶解。
(5)95%乙醇。
(6)硼的标准溶液:准确称取2.858g硼酸,用水溶解定容为1L。该溶液1mL含硼500μg。准确吸取10mL该溶液,加水至1L,再取此溶液10mL,加水至1L,最后所得溶液1mL含硼0.05μg。
3.仪器
分光光度计。
4.方法
(1)取一定量的用无机成分分析用试样的制备干式灰化法(Ⅰ)制备的灰化萃取液中取一定量原液(使其含硼在0.1~20μg),放入瓷制蒸发器中,加一定量的0.1mol氢氧化钙悬浊液,使供试液呈碱性,在水浴上蒸发干固。
(2)放冷后加1mL盐酸-草酸混合液,然后再加0.1%姜黄素溶液2mL混合均匀。
(3)将该混合液在55℃±3℃的水浴上蒸发干固,干固后在相同的温度下保持30min。
(4)放冷后用95%乙醇25mL萃取色素,萃取液用离心分离机分离10min。取其上部清液在530nm波长下测定吸光度。
(5)硼标准溶液也按同样操作,分别对硼含量为0~20μg的标准溶液进行显色测定,制作校正曲线。根据试样的吸光度求出含硼量。
5.计算
按下式计算烟草中硼的含量ρ硼,以10-6mg/kg表示:

式中 mp——被测液含硼量,μg;
V。——灰化萃取原液的体积,mL;
Vt——供显色用被测液量,mL;
m——试样质量,g。
(三十四)烟草中铜的测定——原子吸收光谱法
1.原理
将试样灰化萃取液用原子吸收光谱法分析。
2.试剂和材料
所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
铜的标准溶液:准确称取1g纯铜,加2mL硝酸和6mL盐酸使之溶解,再蒸发干涸,基本干涸后,用水稀释到1L。该溶液含铜1000×10-6mg/kg。用该溶液制成0.2×10-6mg/kg、4×10-6mg/kg、6×10-6mg/kg、8×10-6mg/kg、10×10-6mg/kg的铜的标准溶液。
3.仪器
原子吸收光谱仪,分析线为234.8nm的光源。
4.方法
(1)取一定量的用无机成分分析用试样的制备干式灰化法(Ⅰ)或湿式灰化法制备的灰化萃取原液,如铜含量高时应将其稀释,使铜含量在2×10-6mg/kg~10×10-6mg/kg的范围内。
(2)将铜的标准溶液和待检液用原子吸收光谱法分析。
(3)根据铜的标准溶液的各浓度及其读数做出校正曲线,再从被测液的读数求出被测液中铜的浓度。
5.计算
按下式计算烟草中铜的含量ρ铜,以10-6mg/kg表示:

式中 mp——被测液中铜的浓度,10-6mg/kg;
V——灰化或萃取原液的容量,mL;
d——稀释率;
m——试样质量,g。
(三十五)烟草中镉的测定
1.原理
用二乙基氨荒酸钠和试样湿式灰化萃取液生成螯合物,再用有机溶剂进行液-液萃取,对进入有机溶剂的部分进行原子吸收光谱分析。
2.试剂和材料
所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
(1)酒石酸钾钠溶液:称取25g酒石酸钾钠加水溶解并定容到100mL。
(2)溴酚蓝溶液:称取0.1g溴酚蓝,用20mL乙醇溶解,加水到100mL。
(3)硫酸铵溶液:往水中加硫酸铵直到饱和为止。
(4)二乙基氨荒酸钠溶液:将1g二乙基氨荒酸钠用水溶解并定容到100mL。
(5)甲基异丁基甲酮。
(6)镉的标准原液:准确称取1.000g金属镉,用50mL稀硝酸(浓度60%以上的硝酸)和水按1∶5的比例混合溶解,并煮沸,冷却后加水到1L。
(7)镉的标准溶液:将上述原液用水稀释1000倍。
3.仪器
原子吸收光谱仪,分析线为228.8nm的光源。
4.方法
(1)用无机成分分析用试样的制备湿式灰化法制成灰化萃取液,如用烟末时,1g试样的灰化物用200mL水稀释,不溶的沉淀应尽可能少。
(2)取50mL的试样萃取液放入200mL的分液漏斗中。
(3)加5mL酒石酸钾钠溶液和2滴溴酚蓝指示剂。
(4)加氨水使溶液由淡黄色变成蓝紫色,加水至100mL。
(5)在该溶液中加10mL硫酸铵溶液和5mL二乙基氨荒酸钠溶液。
(6)放置数分钟后加10mL甲基异丁基甲酮,激烈振动5min。
(7)静置后把甲基异丁基甲酮层取出。
(8)空白试验和试样同样处理。
(9)另外镉的标准系列溶液5~20mL及标准溶液的空白试验(与标准溶液含等量硝酸)20mL,也按上述操作同样进行。
(10)用原子吸收光谱仪分析被测液、试样空白试验以及镉的标准系列溶液、标准溶液的空白试验。
(11)根据标准溶液及标准溶液的空白试验的测定值差做校正曲线,再根据试样被测液与空白试验的测定值差,求出被测液中镉的浓度。
5.计算
按下式计算烟草中镉的含量ρ镉,以10-6mg/kg表示:
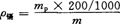
式中 mp——被测液含镉量,10-6mg/kg;
m——试样的质量,g。
(三十六)烟草中铅的测定
1.原理
用试样的湿式灰化萃取原液与吡咯烷荒酸铵生成螯合物,再用有机溶剂进行液体分离,将有机溶剂层进行原子吸收光谱分析。
2.试剂和材料
所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
(1)硫酸铵。
(2)溴酚蓝溶液:称取0.1g溴酚蓝,用20mL乙醇溶解,加水到100mL。
(3)吡咯烷荒酸溶液:称取2.0g吡咯烷荒酸,溶解在100mL水中。
(4)甲基异丁基甲酮。
(5)铅的标准原液:将硝酸铅在100~110℃下干燥,然后准确称取0.16g,溶解在水中,加硝酸10mL后用水定容到1L。
(6)铅的标准溶液:取上述原液25mL,加1mL硝酸后用水定容到500mL。用移液管取该溶液5mL、10mL、20mL、30mL、40mL、50mL,分别放入100mL的容量瓶中。加水到80mL,在加15mL盐酸,用水定容到100mL。该系列溶液每20mL含铅分别为5μg、10μg、20μg、30μg、40μg、50μg。
3.仪器
原子吸收光谱仪,分析线为283.8nm的光源。
4.方法
(1)用无机成分分析用试样的制备湿式灰化法制成灰化萃取液,如用烟末时,1g试样的灰化物用200mL水稀释,不溶的沉淀应尽可能少。
(2)取少于40mL的试样萃取液放入100mL的分液漏斗中。
(3)加20g结晶硫酸铵,基本溶解后加3滴溴酚蓝指示剂。
(4)加氨水稀释,使溶液由淡黄色变成蓝紫色,再加水约至50mL。
(5)加2mL吡咯烷荒酸溶液。
(6)时而振摇一下,放置5min后加10mL甲基异丁基甲酮,激烈振动约5min使充分混合。
(7)静置后把甲基异丁基甲酮层取出,放入带盖的试管中。
(8)空白试验和试样同样处理。
(9)另取20mL溶液,使其含有与标准溶液等量的硝酸及盐酸,作为标准溶液的空白试验。将标准系列溶液及其空白试验溶液各取20mL,分别加10g硫酸铵后,与试样溶液进行同样处理。
(10)用原子吸收光谱仪分析被测液、试样空白试验以及标准系列溶液、标准溶液的空白试验。
(11)用标准溶液及其空白试验的测定值差做校正曲线,再根据试样被测液及其空白试验的测定值差,从校正曲线上求出铅的浓度。
5.计算
按下式计算烟草中铅的含量ρ铅,以10-6mg/kg表示:

式中 mp——被测液含铅量,10-6mg/kg;
m——试样的质量,g。
(三十七)烟草中汞的测定
1.原理
将烟叶用湿式分解法制成试样溶液,再用冷原子吸收分光光度法对还原气产生的水银蒸气进行定量。
2.试剂和材料
所有试剂均应为分析纯,水应为蒸馏水或同等纯度的水。
(1)氯化亚锡溶液:将10g氯化亚锡用20mL温盐酸溶解,加水至100mL。盖上盖放冰箱保存。
(2)硫酸。
(3)汞标准溶液:准确称取0.135g氯化汞,用水溶解,放入1L的容量瓶中。加5mL硝酸后用水定容到1L。每1mL该溶液含100μg汞。准确量取1mL该溶液放入500mL的容量瓶中。加1mL硝酸后用水定容到500mL。每1mL该溶液含0.2μg汞。即为汞标准溶液。
3.仪器
通用型原子吸收分光光度仪或汞专用原子吸收分光光度仪。
汞气化发生装置。
汞中空阴极灯(波长253.7nm)或汞灯。
4.方法
从无机成分分析用试样的制备湿式灰化法制成灰化萃取液中得到并定容(V1)的萃取液中取一定量(100mL以下)作为试样溶液(V2),放入通气试验瓶中,用水调整容量为100mL,连接好装置,再往瓶中加1mL硫酸和2mL氯化亚锡溶液,立刻盖好盖,打开气泵使空气循环,这时记录针急速上升,当达到一定值时测定吸光度。并根据标准溶液绘制的标准曲线求出汞含量。
另取20mL蒸馏水试样按试样湿式分解法进行操作,作为空白试验。并求出空白试验值,它与试样测定值的差即为汞定量值。
5.计算
按下式计算烟草中汞的含量ρ汞,以10-6mg/kg表示:

式中 mp——被测液汞含量,μg;
m——试样的质量,g;
V1——灰化萃取原液的体积,mL;
V2——供显色被测液量,mL。
(三十八)烟草pH值的测定
1.试剂和材料
(1)蒸馏水。
(2)缓冲溶液。
2.仪器
万分之一分析天平。
pH计。
500mL锥形烧瓶。
电磁搅拌器。
3.试样的称量
相当于5g干粉质量的粉状烟叶的质量=5.000×100
式中 100——粉状烟叶的湿度百分值。
4.方法
(1)校准天平的零点。
(2)校准pH计。
(3)将相当于5g干粉质量的粉状烟叶倒入500mL锥形烧瓶中,加入50mL蒸馏水,盖上瓶塞。用电磁搅拌器搅拌15min。
(4)停止搅拌并量度悬浮液的pH,精确至0.01pH单位。