颅脑创伤患者过度通气的应用
出处:按学科分类—医药、卫生 第二军医大学出版社《颅脑创伤临床救治指南》第87页(16761字)
简介
1.结论
在没有颅内压(ICP)增高的情况下,应避免将长时程过度通气疗法(PaCO2≤3.3kPa)应用于治疗重型脑创伤(TBI)患者。
2.原则
在重型TBI后最初24h应避免使用预防性过度通气疗法,因为它在脑血流(CBF)减少的情况下能进一步降低脑灌注压。
3.方法选择
当使用镇静剂、肌松剂、脑脊液(CSF)引流和渗透性利尿剂难以控制颅内高压,脑受压所致的脑功能障碍进行性加重时,短暂过度通气疗法可能是有益的。
如果必需使用短暂过度通气,颈静脉氧饱和度(SjO2)、动-静脉氧含量差(A-VDO2)和脑血流量(CBF)监测有助于识别由于过度通气所致的PaCO2<4.0kPa而引起的脑缺血。脑组织氧直接测定技术更能直接反映脑组织含氧量。
一、概述
强制性过度通气(PaCO2≤3.3kPa)在20多年时间里是处理严重TBI后颅内高压的基本方法,因为它会使ICP迅速下降。然而,至今没有研究表明,过度通气能改善严重TBI患者预后。40%的严重TBI患者出现脑肿胀和ICP升高;TBI后ICP增高是引起死亡和脑功能障碍的最常见原因之一。因此,大多数临床医师设想通过过度通气降颅压来改善所有重型TBI患者的预后。
然而,过度通气是通过引起脑血管收缩和CBF减少来降低ICP。过去20年中研究确实证明了CBF在伤后第1天低于正常值的一半,但强制性过度通气有造成脑缺血的危险。在大多数严重TBI死亡病例中都发现了存在脑缺血的组织学证据。一组前瞻性随机研究发现不使用预防性过度通气和使用过度通气相比,伤后3个月和6个月随访发现不使用过度通气的患者预后较使用者更好。因此,限制重型TBI后过度通气的应用则可能有助于改善脑神经功能恢复以及避免医源性脑组织缺氧。
二、论点形成过程
上述作者首先对过去25年中所有发表的相关文章进行广泛回顾。通过计算机检索国家医学图书馆,得到大约400篇文章。检索用下列医学主题词:脑创伤、脑缺血、颈静脉、局部脑血流、脑灌注、脑过度灌注。综述了相关文章写成初稿。作者尤其重视颅脑伤后CBF、A-VDO2、SjO2和过度通气。所有这些文章都是8个病例以上的临床随机化研究,仅1篇是双盲随机对照前瞻性临床研究。
三、科学基础
(一)CBF与重型TBI
CBF在伤后最初24h内最低,并在随后3d中逐渐增加,除难以控制性颅内高压所致死亡患者外。典型重型颅脑伤患者的CBF在伤后最初8h内<30ml/(100g·min),且在伤后最初4h内可能<20ml/(100g·min)。
TBI后引起不可逆性缺血或梗死的CBF阈值尚不清楚。Obrist等提出TBI会引起脑代谢抑制,因此,TBI后CBF减少在多数情况下可能与脑代谢相适应。然而在一组正电子发射断层扫描(PET)研究中,Heiss等观察到16例临床和CT证明的半球卒中患者在症状出现后平均23h,梗死中心的CBF平均为(16.7±7.95)ml/(100g·min),梗死灶附近区域CBF为(31.0±10.65)ml/(100g·min)。
严重TBI时,CBF在硬膜下血肿、弥散性损伤和低血压患者中最低,在硬膜外血肿或CT扫描正常的患者中最高。在伤后最初24h内CBF与GCS评分或预后有直接的相关。CBF与ICP变化并不始终呈平行关系,在某些病例中,CBF升高会引起ICP降低。
(二)脑血管的CO2调节机制
脑细小动脉的管径受到动脉血CO2分压(PaCO2)的调节,这种调节机制对于脑血管阻力(cerebral vascular resistanc,CVR)、CBF以及脑血容量(cerebral blood volume,CBV)具有重要意义。PaCO2的降低引起细小动脉收缩,CVR增加,CBF相应减少;PaCO2的升高则作用相反。实际上CO2的调节作用是通过脑脊液pH值的变化来实现的:pH值降低,血管扩张;pH值升高,血管收缩。
血和脑脊液中的pH值主要取决于缓冲对HCO3-/H2CO3的比值。CO2和H2CO3以及 之间存在以下关系:
之间存在以下关系:
过度通气时全身的PaCO2降低,pH值增高,数小时至数天后,受到肾脏调节作用,血 浓度亦降低,血pH值逐渐代偿接近正常水平。因为CO2能够自由通过血脑屏障,过度通气同样引起脑脊液PaCO2的迅速降低和pH值的相应增高,但由于脉络丛碳酸酐酶的作用,
浓度亦降低,血pH值逐渐代偿接近正常水平。因为CO2能够自由通过血脑屏障,过度通气同样引起脑脊液PaCO2的迅速降低和pH值的相应增高,但由于脉络丛碳酸酐酶的作用,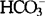 浓度在数小时内很快降低,而H+和
浓度在数小时内很快降低,而H+和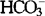 不能自由通过血脑屏障,故脑脊液pH值较快得以代偿。这种变化特点的临床意义在于,过度通气通过降低血和脑脊液中的PaCO2,脑脊液的pH值相应增高,引起脑血管收缩、CBF降低,CBV相应减少,达到降低ICP的效果。但在数小时内,脑脊液的pH值便因为脉络丛碳酸酐酶的作用得到代偿,达到或接近过度通气前的水平,脑细小动脉的管径、CBF、CBV以及ICP亦随之恢复到初始水平。因此,过度通气难以较长时间地维持降颅压的作用;而且,过度通气一旦终止,脑脊液中PaCO2相对于
不能自由通过血脑屏障,故脑脊液pH值较快得以代偿。这种变化特点的临床意义在于,过度通气通过降低血和脑脊液中的PaCO2,脑脊液的pH值相应增高,引起脑血管收缩、CBF降低,CBV相应减少,达到降低ICP的效果。但在数小时内,脑脊液的pH值便因为脉络丛碳酸酐酶的作用得到代偿,达到或接近过度通气前的水平,脑细小动脉的管径、CBF、CBV以及ICP亦随之恢复到初始水平。因此,过度通气难以较长时间地维持降颅压的作用;而且,过度通气一旦终止,脑脊液中PaCO2相对于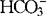 迅速恢复正常,pH值相应降低,引起脑血管的扩张,CBF、CBV以及ICP出现反弹甚至超过通气前的水平。在动物实验中发现,过度通气24h后,回升PaCO2引起脑血管的明显扩张,血管口径超出过度通气前的大小;在人体研究中观察到,维持PaCO2在2.0~2.7kPa水平4h后,CBF恢复至接近原来水平,5h后终止过度通气,CBF出现明显增高并超出原先的31%。
迅速恢复正常,pH值相应降低,引起脑血管的扩张,CBF、CBV以及ICP出现反弹甚至超过通气前的水平。在动物实验中发现,过度通气24h后,回升PaCO2引起脑血管的明显扩张,血管口径超出过度通气前的大小;在人体研究中观察到,维持PaCO2在2.0~2.7kPa水平4h后,CBF恢复至接近原来水平,5h后终止过度通气,CBF出现明显增高并超出原先的31%。
(三)颅脑损伤后脑血流和脑代谢水平的改变
过度通气在减少CBF、降低CIP的同时,有引起脑缺血损害的危险,尤其是在伤后24h内或过分使用过度通气的情况下。这个问题涉及到颅脑损伤后CBF和脑代谢水平的改变。脑缺血损害取决于CBF和脑代谢水平之间的平衡。当CBF降低不能满足脑组织的代谢需求时,即发生脑缺血性损害。如果在重型TBI后业已存在脑组织的缺血,过度通气无疑会加重这一病理过程。
对重型TBI患者的尸解组织切片中发现,缺血性脑损伤普遍存在。但这种缺血性脑损伤是发生在受伤当时,还是继发于伤后CBF的减少,尚不能肯定。研究发现重型TBI后的早期,CBF明显减少。Marion等报道重型TBI后1~4h时,CBF平均为(27±14)ml/(100g·min),伤后5~24h升至(44±10)ml/(100g·min)。Bouma等报道的伤后6h内CBF为(22.5±5)ml/(100g·min),6h后CBF开始增加,36~42h达到高峰。尽管上述发现证实重型TBI后24h内存在CBF的降低,但并不能说明脑组织有缺血性损害,因为脑组织缺血与否除与CBF有关外,还与脑组织的代谢水平密不可分。
当脑血流减少时,为满足代谢需要,脑组织会增加对血流中氧的提取,氧提取分数可由正常状态下的30%~40%升高到90%,A-VDO2增大。当仍不足以代偿时,脑组织的能量代谢便出现障碍。正常脑代谢状况下,CBF低于20ml/(100g·min)便会出现神经电活动的异常,低于15~18ml/(100g·min)即出现脑缺血性损害。但是当脑组织的代谢水平降低,如使用巴比妥药物后,脑需氧量相应减少,脑组织对缺血的耐受性增加,CBF还会出现继发性的下调,称之为CBF的代谢自动调节作用。Obrist等发现重型TBI后患者在CBF降低的同时,有脑组织的氧代谢率(CMRO2)的降低,而氧提取分数正常,A-VDO2在正常范围以内,提示CBF的降低是继发于脑组织代谢水平的下降,而并不支持脑缺血性损害的变化。此外的研究亦证实,在重型TBI患者,无论CBF降低抑或是升高,CMRO2均降低,且CMRO2水平与患者的意识和预后相关。
综上所述,在重型TBI后的早期24h以内,存在CBF的降低,但目前尚不能证实此时的CBF降低伴有脑缺血性损害,CBF降低可能是继发于伤后脑组织代谢水平下降的一种改变。尽管如此,对于过度通气的运用来说,伤后早期可能使已降低的CBF更加降低,有加重或引起脑缺血损害之虞。
(四)过度通气对CBF的影响
动物实验发现,当过度通气PaCO2达到1.3~2.0kPa时,CBF下降至18ml/(100g·min);当PaCO2降至3.2~3.5kPa时,CBF下降的同时A-VDO2增高,脑组织的CMRO2不变,提示脑组织通过增加对血流中氧的提取以代偿CBF的下降,维持脑组织的能量代谢;但不适度的过度通气还是会引起神经元电活动的异常和CMRO2的下降,尤其当ICP增高接近引起脑缺血的阈值时,过度通气使PaCO2达到2.0kPa,即出现脑缺氧变化。
在正常人体研究发现,当过度通气使颈内静脉氧分压降至2.8kPa时,即出现脑电图改变。Cold等在临床观察到,过度通气前就存在CBF降低的重型TBI患者,在过度通气后脑内CBF降低区域的范围发生扩大,但因为没有同时检测脑代谢率,不能判断CBF的下降是否导致脑缺血;Obrist在一部分具有较高CMRO2而CBF较低的重型TBI患者中发现,当过度通气使PaCO2降至3.1kPa时,CBF下降到19ml/(100g·min),脑A-VDO2增大,CMRO2下降近1/2,提示脑能量代谢受到影响,此时颈内静脉氧分压从5.0kPa降至3.0kPa。
上述研究表明,过度通气在部分患者,尤其是在已有CBF降低的患者,可能会导致脑组织缺血和能量代谢障碍。只是在人体引起脑组织缺血的阈值目前尚不确切。
(五)A-VDO2和SjO2
除硬膜下血肿患者外,A-VDO2与CBF在伤后24h内呈负相关。A-VDO2增加9%容积可能提示脑缺血。
SiO2通常大于50%,低于50%被认为饱和度下降。长时程和饱和度降低与患者预后不良有关。饱和度下降常出现CBF下降。低碳酸血症与饱和度下降相关。在6例患者中,Cruz等发现当动脉PaCO2<2.9kPa时,SjO2平均为45%±8%,当PaCO2增加1.3kPa后,SjO2为59%±3.2%。Sheinberg等证明PaCO2<3.7kPa是33例患者中10例饱和度下降的原因。
(六)过度通气
过度通气能有效降低ICP的前提是脑血管具有对CO2的反应性。尽管在部分重型TBI终末期的患者,脑血管丧失了对CO2的反应性,但在多数情况下,这一反应性仍然存在。
1.预防性持续过度通气
对于预防性持续过度通气的运用尚存在争议。一组研究将重型TBI患者(GCS≤8分)随机分为三组:正常通气组、过度通气组、过度通气+tromethamine(THAM,一种纠正酸中毒的氨基缓冲剂)组,后两组持续应用过度通气5d。结果在GCS运动评分4~5分的患者中发现,过度通气组患者的预后不如另外两组;同时发现,过度通气后的24h和72h,联合应用THAM的过度通气组中,CBF较基线下降25%,而单用或未用过度通气组的CBF与基线比较没有变化。通过此研究作者得出结论,预防性持续过度通气在部分重型TBI患者具有不利影响,而联合静脉应用THAM能够克服这一不利影响。THAM是一种可通过血脑屏障的弱碱性药物。过度通气后的几个小时,脉络丛通过降低CSF中 浓度,使脑脊液pH值很快恢复,当静脉应用THAM后,因其缓冲作用使CSF的pH值维持在较高水平,同时也有利于过度通气对伤后脑组织乳酸酸中毒的缓解作用。此外,Obrist等观察到,在部分重型TBI患者的伤后4d内,CBF不是降低而是升高,这种高血流状态平均持续3d,在伤后24h即可出现,并且与ICP增高密切相关;而CBF对过度通气的反应在这些患者中更为明显。
浓度,使脑脊液pH值很快恢复,当静脉应用THAM后,因其缓冲作用使CSF的pH值维持在较高水平,同时也有利于过度通气对伤后脑组织乳酸酸中毒的缓解作用。此外,Obrist等观察到,在部分重型TBI患者的伤后4d内,CBF不是降低而是升高,这种高血流状态平均持续3d,在伤后24h即可出现,并且与ICP增高密切相关;而CBF对过度通气的反应在这些患者中更为明显。
上述发现提示在重型TBI的治疗中,可能存在一个适用预防性持续过度通气的窗口期,同时联合应用一些弱碱性药物如THAM,可维持过度通气的降压作用,消除其对预后的不利影响。
2.过度通气在顽固性颅内高压中的应用
目前过度通气的临床运用得到认可的是作为一种临时的救治手段,在ICP急剧增高或难以利用其他降颅压措施控制时,可予以合理使用。
由于ICP的波动性,在颅脑损伤的患者,ICP会出现短暂的自发性增高,或者由于各种刺激因素如吸痰、体位变动等骤然增高。虽然这种一过性的变化历时不过数分钟,但可导致脑灌注压的急剧下降。在这种情况下,可短时应用过度通气,有利于缓解ICP增高。在一过性因素消除后,即可停止过度通气。这种短时过度通气的应用尚不致引起脑脊液中 的变化,故不存在ICP的反跳现象。
的变化,故不存在ICP的反跳现象。
对于一些颅脑损伤的患者出现难以控制的颅内高压,当其他降颅压措施如脑室外引流、手术清除血肿或内、外减压仍不能有效控制,或者还来不及采取这些处理时,可使用过度通气作为救治手段。然而,这种情况下一旦用上了过度通气,因为停用后的反弹现象,可能难以撤除。
Raichle等研究了一组健康志愿者对过度通气的正常反应:PaCO2降至2.0~2.7kPa30min后,CBF减少了40%;4h后CBF增加到基础值的90%。当PaCO2恢复正常后,CBF超过正常值31%。
在重型TBI中CO2相关的血管反应性为:PaCO2每变化0.13kPa,CBF变化3%。但在CBF较低时变化值较小。血管对CO2反应性降低与预后不良相关。在很多患者中,局部CO2血管反应性与全脑血管反应性相差大于50%。在部分患者中,脑自主调节在正常碳酸血症时保存,而在低碳酸血症时丧失。在某些病例,过度通气确实可造成ICP的下降。Crockard等发现14例患者中4例PaCO2下降至3.3~4.0kPa,ICP也相应降低。Obrist等发现31例患者中仅15例出现过度通气后ICP下降,但有29例出现CBF下降。强制性过度通气能造成A-VDO2和CBF降到或接近脑缺血阈值。在10例PaCO2为(3.1±0.4)kPa的患者中,Obrist等发现A-VDO2为(10.5±0.7)容量%,CBF为(18.6±4.4)ml/(100g·min)。
1991年,Muizelaar等发表了一组随机化前瞻性临床研究结果。此研究将77例严重TBI患者随机分为两组,一组伤后5d内以长时程过度通气[PaCO2为(3.3±0.4)kPa]治疗,一组在同一时期内保持相对正常血碳酸值[PaCO2为(4.7±0.3)kPa]。在伤后3个月和6个月随访,过度通气组患者预后明显差于正常血碳酸组。伤后1年随访未发现两组患者预后有明显统计学差异。然而,差异的不明显可能由于统计学错误造成,因为伤后1年实际上只有较少患者得到随访。
3.过度通气使用方法和过度通气的终止
利用过度通气控制颅压的患者多处于昏迷状态,需要气管插管维持呼吸道的通畅,同时也有利于机械辅助过度通气的应用。气管插管可引起血压和ICP的急剧增高,诱发脑疝形成,故插管前需静脉用药,如硫喷妥钠、利多卡因或依托咪酯能缓解ICP的增高。
要达到降低PaCO2的目的,需要提高每分钟通气量(潮气量×呼吸频率)。通常利用增加呼吸频率来提高每分钟通气量,而维持正常潮气量不变(10~12ml/kg),这样能避免增加潮气量引起胸腔压的增高,进而导致ICP升高。常用的观察指标为呼气末CO2分压。由于肺内通气/血流灌注比的不均衡和气道无效腔对呼出CO2的稀释,呼气末CO2分压稍低于PaCO2,如果能行血气分析明确呼气末CO2分压和PaCO2的相关关系,可更好地实现对PaCO2的监测。一般PaCO2控制在2.7~4.0kPa的范围,同时需要对CMRO2、CBF、A-VDO2等指标的观察,以防止出现脑缺血损害。
过度通气的终止应循序渐进。过度通气的终止对ICP的影响取决于两个因素,即过度通气时间的长短和当时颅内容物的顺应性。短暂使用过度通气的患者,在ICP得到控制后即可终止,不必担心ICP的反弹。而对于较长时间应用过度通气的患者,过度通气的终止应在ICP变化的监测下,遵循个体化原则。开始时可试行将呼吸频率减少1~2次,在有些患者即可见到ICP立即升高,如果升高不显着,维持该频率直到CSF经重新调节适应后引起ICP回降,再依据这样的变化规律进行以后的调整;如果颅内压在呼吸频率降低1~2次后显着升高,说明此时颅内的顺应性很差,需综合采用其他降低颅内压的措施后,才可能安全地终止过度通气。
目前我国尚没有统一的过度通气临床使用准则。美国《重型颅脑创伤救治指南》提出的过度通气使用原则为:当重型TBI后存在CBF的降低时,过度通气会进一步影响脑组织的血流灌注,因此重型TBI后的24h内应避免预防性地使用过度通气(PaCO2≤4.7kPa);当神经症状急剧恶化时,可考虑短时使用过度通气;对于顽固性的颅内高压,在镇静、脱水、CSF引流等治疗措施控制无效时,可较长时程使用过度通气。SjO2、A-VDO2、CBF等指标的监测有助于对脑缺血的判断和预防。
四、小结
长时程过度通气疗法应被禁止用于重型rBI后最初5d,特别是最初24h。重型TBI患者的CBF监测证明伤后早期脑血流降低。过度通气能进一步降低CBF,但并不肯定能降低ICP,且可能造成脑血管自主调节功能变化。在严重脑损伤患者,脑血管对低碳酸血的反应降低,且灌注压下降。虽然尚未确立发生不可逆脑缺血的CBF水平,但在TBI后死亡患者中90%被证明有缺血性细胞损害改变。PET检查也证明不可逆性脑损害可能发生于CBF降至<15~20ml/(100g·min)时。随机化前瞻性临床试验表明,长时程过度通气疗法治疗不但不能提高重型TBI患者预后,而且会增加患者的死亡率。
五、前景与展望
仍然需要更多大宗随机化前瞻性临床试验,以确定伤后最初24h内的短程过度通气是否有害。
六、主要依据
支持本章观点作者的研究概要及结论见表11-1、11-2和11-3。
表11-1 重型TBI后CBF的临床测量

表11-2 TBI后缺血的组织学证据和伤后A-VDO2增宽的证据

表11-3 过度通气对CBF、A-VDO2、SjO2和临床预后的影响

【参考文献】:
1 Becker DP,Miller JD,Ward JD,et al.The outcome from severe head injury with early diagnosis and intensive management.J Neurosurg,1977,47:491
2 Bouma GJ,Muizelaar JP.Relationship between cardiac output and cerebral blood flow in patients with intact and with impaired autoregulation.J Neurosurg,1990,73:368
3 Bouma GJ,Muizelaar JP,Choi SC,et al.Cerebral circulation and metabolism after severe traumatic brain injury:the elusive role of ischemia.J Neurosurg,1991,75:685
4 Bouma Gi,Muizelaar JP,Stringer WA,et al.Ultra-early evaluation of regional cerebral blood flow in severely head injured patients using Xenon enhanced computed tomography.J Neurosurg,1992,77:360
5 Bauma GJ,Muizelaar JP,Stringer WA,et al.Ultra-early evaluation of regional cerebral blood flow in severely head-injured pationts using Xenon-enhanced computerized tomography.J Neurosurg,1993,77(3):360
6 Cold GE.The relationship between cerebral metabolic rate of oxygen and cerebral blood flow in the acute phase of head injury.Acta Anaesthesiol Scand,1986,30:453
7 Cold GE.Measurements of CO2 reactivity and barbiturate reactivity in patients with severe head injury.Acta Neurochir(Wien),1989,98:153
8 Cold GE,Christensen MS,Schmidt K.Effect of two levels of induced hypocapnia on cerebral autoregulation in the acute phase of head injury coma.Acta Anaesthesiol Scand,1981,25:397
9 Crockard HA,Coppel DL,Morrow WF.Evaluatoon of hyperventilation in treatment of head injuries.Br Med J,1973,4:634
10 Cruz J.On-line monitoring of global cerebral hypoxia in acute brain injury.Relationship to intracranial hypertension.J Neurosurg,1993,79:228
11 Cruz J.Low clinical ischemic threshold for cerebral blood flow in severe acute brain trauma:Case re port.J Neurosurg,1994,80:143
12 Cruz J,Miner ME,Allen SJ,et al.Continuous monitoring of cerebral oxygenation in acute brain injury:assessment of cerebral hemodynamic reserve.Neurosurgery,1991,29:743
13 Diringer MN,Yundt K,Videen TO,et al.No reduction in cerebral metabolism as a result of early moderate hyperventilation following severe traumatical brain injury.J Neurosurg,2000,92(1):7
14 Enevoldsen EM,Jensen FT.Autoregulation and CO2 responses of cerebral blood flow in patients with acute severe head injury.J Neurosurg,1978,48(5):689
15 Fieschi C,Battistini N,Beduschi A,et al.Regional cerebral blood flow and intraventricular pressure in acute head injuries.J Neurol Neurosurg Psychiatry,1974,37:1378
16 Geraci E,Geraci T.A look at recent hyperventilation studies:outcomes and recommendations for early use in the head-injured patient.J Neurosci Nurs,1996,28(4):222
17 Gotoh F,Meyer JS,Takagi Y.Cerebral effects of hyperventilation in man.Arch Neurol,1965,12:410
18 Graham DI,Adams JH.Ischaemic brain damage in fatal head injuries.Lancet,1971,1:265
19 Graham DI,Lawrence AE,Adams JH,et al.Brain damage in fatal non-missile head injury without high intracranial pressure.J Clin Pathol,1988,41:34
20 Heiss WD,Huber M,Fink GR,et al.Progressive derangement of peri-infarct viable tissue in ischemic stroke.J Cereb Blood Flow Metab,1992,12:193
21 Imberti R,Ciceri M,Bellinzona G,et al.The use of hyperventilation in the treatment of plateau waves in two patients with severe traumatic brain injury:comtrasting effects on cerebral oxygenation.J Neurosurg Anesthesiol,2000,12(2):124
22 Jaggi JL,Obrist WD,Gennarelli TA,et al.Relationship of early cerebral blood flow and metabolism to outcome in acute head injury.J Neurosurg,1990,72:176
23 Kent DY,Michael ND.The use of hyperventilation and its impact on cerebral ischemia in the treatment of traumatic brain injury.Crit Care Clin,1997,13(1):163
24 Marmarou A.Intracellular acidosis in human and exoerimental brain injury.J Neurotrauma,1992,9(Suppl 2):s551
25 Marion DW,Bouma GJ.The use of stable Xenon-enhanced computed tomographic studies of cerebral blood flow to define changes in cerebral carbon dioxide vasoresponsivity caused by a severe head injury.Neurosurgery,1991,29:869
26 Marion DW,Darby J,Yonas H.Acute regional cerebral blood flow changes caused by severe head injuries.J Neurosurg,1991,74:407
27 Marshall LF,Smith RW,Shapiro HM.The outcome with aggressive treatment in severe head injuries.Part 1.The significance of intracranial pressure monitoring.J Neurosurg,1979,50:20
28 Miller JD,Becker DP,Ward JD,et al.Significance of intracranial hypertension in severe head injury.J Neurosurg,1977,47:503
29 Muizelaar JP,Marmarou A,DeSalLes AA,et al.Cerebral blood flow and metabolism in severely head-inuryed children.Part 1:Relationship with GCS score,outcome,ICP,and PVI.J Neurosurg,1989,71:63
30 Muizelaar JP,Marmarou A,Ward JD,et al.Adverse effects of prolonged hyperventilation in patients with severe head injury:a randomized clinical trial.J Neurosurg,1991,75:731
31 Narayan RK,Kishore PRS,Becker DP,et al.Intracranial pressure:to monitor or not to monitor.J Neurosurg,1982,56:650
32 Obrist WD,Gennarelli TA,Segawa H,et al.Relation of cerebral blood flow to neurological status and outcome in head-injured patients.J Neurosurg,1979,51:292
33 Obrist WD,Langfitt TW,Jaggi JL,et al.Cerebral blood flow and metabolism in comatose patients with acute head injury.J Neurosurg,1984,61:241
34 Raichle ME,Plum F.Hyperventilation and cerebral blood flow.Stroke,1972,3:566
35 Raichle ME,Posner JB,Plum F.Cerebral blood flow during and after hyperventilation.Arch Neurol,1970,23:394
36 Robertson CS,Clifton GL,Grossman RG,et al.Alterations in cerebral availability of metabolic substrates after severe head injury.J Trauma,1988,28:1523
37 Robertson CS,Contant CF,Gokaslan ZL,et al.Cerebral blood flow,arteriovenous oxygen difference,and outcome in head injured patients.J Neurol Neurosurg Psychiatry,1992,55:594
38 Ross DT,Graham DI,Adams JH.Selective loss of neurons from the thalamic reticular nucleus following severe human head injury.J Neurotrauma,1993,10:151
39 Salvant JB,Muizelaar JP.Changes in cerebral blood flow and metabolism related to the presence of subdural hematoma.Neurosurgery,1993,33:387
40 Schroder ML,Muizelaar JP,Kuta AJ.Documented reversal of global ischemia immediately after removal of an acute subdural hematoma.Neuroscugery,1994,80:324
41 Sheinberg M,Kanter MJ,Robertson CS,et al.Continuous monitoring of jugular venous oxygen saturation in head-injured patients.J Neurosurg,1992,76:212
42 Tenjin H,Yarnaki T,Nakagawa Y,et al.Impairment of CO2 reactivity in severe head injury patients:an investigation using thermal diffusion method.Acta Neurochir(Wien),1990,104:121
43 Thiagarajan A,Goverdhan PD,Chari P,et al.The effect of hyperventilation and hyperoxia on cere bral venous oxygen saturation in patients iwth traumatic brain injury.Anesth Analg,1998,87(4):850
44 Wolf AL,Levi L,Marmarou A,et al.Effect of THAM upon outcome in severe head injury:a randomized prospective clinical trial.J Neurosurg,1993,78(1):54
45 Yoshida K,Marmarow A.Effects of tromethamine and hyperventilation on brain injury in the cat.J Neurosurg,1991,74:87