增塑剂的检验
出处:按学科分类—工业技术 广东经济出版社;中国轻工业出版社《烟草工业手册》第717页(25110字)
(一)取样
(1)从每批桶数的5%中选取试样,小批者不少于5桶,用玻璃取样管自每桶中取出均匀的试样,总体积不小于1000mL,收集于清洁干燥的磨口瓶中,混匀。
(2)将所取试样分别装于两个清洁干燥的磨口瓶中,瓶口加封并注明生产厂名、产品名称、规格、取样日期、取样者,一瓶作分析,另一瓶保存备查。
(二)外观色泽测定
1.铂-钴比色法
(1)仪器:
比色管:磨口无色比色管,100mL、50mL。
比色箱及比色架,见图8-2-30。

图8-2-30 比色箱及比色架
烧杯:150mL。
容量瓶:1000mL(棕色磨口)。
(2)试剂:氯化钴、氯铂酸钾、盐酸、蒸馏水、二次蒸馏水。
(3)三种500号铂-钴比色标准原液的配制:
①黄色:准确称取1.2450g氯铂酸钾和1.0000g氯化钴(准确至0.0002g),溶于100mL盐酸中,然后用蒸馏水稀释至1000mL。此溶液即为500号色度标准比色液(I)。
②黄中带绿:准确称取1.5000g氯铂酸钾和0.3000g氯化钴(准确至0.0002g),溶于100mL盐酸中,然后用蒸馏水稀释至1000mL。此溶液即为500号色度原液(Ⅱ)。
③黄中带红:准确称取0.8000g氯铂酸钾和2.0000g氯化钴(准确至0.0002g),溶于100mL盐酸中,然后用蒸馏水稀释至1000mL。此溶液即为500号色度标准溶液(Ⅲ)。
④取不同量的500号原液,用0.1mol/L盐酸溶液稀释至100mL即可制得任意号数的色度标准,其计算式如下:
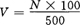
式中 V——配制100mLN号色度标准溶液所取500号标准原液的体积,mL;
N——欲配制的色度标准液的号数。
500号原液应贮存于棕色瓶中,置暗处保存,有效期6个月。稀释的N号铂-钴比色液,应贮存于具有磨口塞的比色管中,比色后置于暗处存放,有效期为1个月。
(4)方法:将试样均匀混合后,注入试管中,用肉眼观察,应是透明液体,无混浊现象,无明显机械杂质,在室温条件下进行比色测定。
取两支颜色相同,高度相等的比色管,一支注入100mL或50mL试样,另一支注入相同体积的色度标准液,置于装有反光镜的比色架上,拿下比色管盖,将此比色架放入比色箱中,转动反光镜,以反光镜中反射的颜色进行比色。
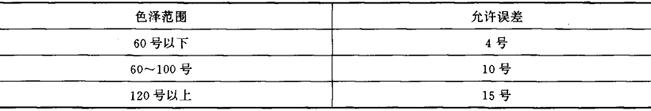
平行测定结果的允许误差应符合表8-2-14的规定。
表8-2-14 色泽范围与允许误差
2.碘比色法
(1)仪器:
比色管:磨口无色比色管,100mL、50mL。
比色箱及比色架,见图8-2-30。
烧杯:200mL。
容量瓶:1000mL(棕色磨口)。
试剂瓶:1000mL(棕色磨口)。
(2)试剂:
碘、碘化钾、蒸馏水:二次蒸馏水。
(3)碘比色标准原液的配制:
①将碘置于硫酸干燥器中,干燥48h。
②向装有10mL已溶55g碘化钾的二次蒸馏水的容量瓶中,加入干燥的碘1.0000g(准确至0.0002g),充分溶解后,用蒸馏水稀释至1000mL。此溶液为100mg碘/100mL碘化钾,定为标准溶液100号。放置2~3d方可使用。
③取不同体积的100号标准溶液,用蒸馏水稀释至PmL,即可制得任意号数的色度标准,其计算式如下:
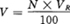
式中 V——配制PmLN号色度标准溶液所取100号标准原液的体积,mL;
N——欲配制的色度标准液的号数;
VR——所用比色管的毫升数。
100号碘标准原液(密封保存于棕色磨口瓶中)及稀释的碘标准比色液都要置暗处存放。有效期为15d。
(4)方法:将试样均匀混合后,注入试管中,用肉眼观察,应是透明液体,无混浊现象,无明显机械杂质,在室温条件下进行比色测定。
取两支颜色相同,高度相等的比色管,一支注入100mL或50mL试样,另一支注入相同体积的色度标准液,置于装有反光镜的比色架上,拿下比色管盖,将此比色架放入比色箱中,转动反光镜,以反光镜中反射的颜色进行比色。
平行测定的两个结果差数不应超过0.5mg碘/100mL碘化钾(即0.5号)。
以平行测定两个结果的算术平均值作为试样的色泽。
(三)折光率测定
1.仪器
阿贝型折光仪。恒温水浴:准确度0.5℃。
2.方法
将折光仪放在光线充足的位置,与恒温水浴连接好,将折光仪棱镜的温度调节至20℃,分开两面棱镜于20℃保持数分钟,调节棱镜的螺旋至视场分为明暗两部分,转动补偿器消除彩虹并使明暗分界线清晰。继续调节螺旋使明暗分界线对准在十字线上。根据标尺刻度记录读数。读数应准确至小数点后第四位(最后一位为估计数字)。轮流从一边再从另一边将分界线对准在十字线上,重复观察及记录读数3次,读数间的差数不得大于0.0003。所得读数的平均值即为试样的折光率。
折光仪使用前应用水校正,20℃时水的折光率为1.3330。
(四)相对密度测定
1.相对密度瓶法
(1)原理:在20℃时,分别测定充满同一相对密度瓶的水及试样的质量,由水的质量可确定相对密度瓶的容积即试样的体积,根据试样的质量及体积可计算其相对密度。
(2)仪器:
分析天平:感量为0.1mg。
相对密度瓶:是一个带有侧管,尖端是毛细管状,中间为一带有玻璃磨口温度计的小瓶,温度计的分度值为0.2℃,瓶的容积为15~25mL(见图8-2-31)。

图8-2-31 相对密度瓶
1-相对密度瓶 2-侧管 3-侧孔 4-罩 5-温度计
恒温水浴:准确度0.1℃。
(3)方法:将相对密度瓶洗净并干燥,带温度计及侧孔罩称量。然后取下温度计及侧孔罩称量,用新煮沸并冷却到15℃的蒸馏水充满相对密度瓶,不得带入气泡,插入温度计,将相对密度瓶置于20.0℃±0.1℃的恒温水浴中,至相对密度瓶温度计达20℃,并使侧管中的液面与侧管口齐平,立即盖上侧孔罩,取出相对密度瓶,用滤纸擦干其外壁上的水,立即称量。
将相对密度瓶内的水倒出,洗净并使之干燥,带温度计及侧孔罩称量。然后用试样代替水重复上述操作。
(4)计算:
试样在20℃时的相对密度按下式计算:

式中 ρ——试样在20℃时的相对密度,g/mL;
m1——20℃时充满相对密度瓶所需试样的表观质量,g;
m2——20℃时充满相对密度瓶所需蒸馏水的表观质量,g;
ρ0——20.0℃时蒸馏水的相对密度(=0.99820g/mL);
A——空气浮力校正值;
ρa——干燥空气在20℃,1013.25hPa时的相对密度值(≈0.0012g/mL)。
2.韦氏天平法
(1)原理:在20℃时,分别测量浮锤在水及试样中的浮力。由于浮锤所排开水的体积与所排开试样的体积相同,所以,根据水的相对密度及浮锤在水及试样中的浮力即可计算出试样的相对密度。
(2)仪器:见图8-2-32。

图8-2-32 韦氏天平原理图
1-支架 2-调节器 3-指针 4-横梁 5-刀口 6-游码 7-小钩 8-细白金丝 9-浮锤 10-玻璃简 11-调整螺丝
浮锤内温度计的分度值为0.1℃。
恒温水浴:准确度0.1℃。
(3)方法:见图8-2-32所示安装好韦氏天平。将浮锤悬挂在小钩上,如果两个指针没有互相对正,则旋转调整螺丝,使两个指针对正为止。
向玻璃筒中注入刚煮沸并冷却至20.0℃±0.1℃的水,然后将浮锤全部浸入水中。将玻璃筒置于20.0℃±0.1℃的恒温水浴中,静置15~20min后,把单位游码挂在小钩上。这时天平应保持平衡。
将玻璃筒中水倾出,玻璃筒及浮锤先用乙醇后用乙醚洗涤数次吹干。注入预先调整至20.0℃±0.1℃的试样。同时置于20.0℃±0.1℃的恒温水浴中,静置15~20min后,调整游码,使梁上游码都放在刻度上,依据从大游码到小游码调整游码至天平保持平衡。记录读数。
将浮锤取出,使其完全干燥,在相同温度下,用试样代替水重复以上操作。
(4)计算:

式中 ρ——试样在20℃时的相对密度,g/mL;
ρ1——浮锤浸于水中时游码的读数;
ρ2——浮锤浸于试样中时游码的读数;
ρ0——20.0℃时蒸馏水的相对密度(=0.99820g/mL)。
(五)沸点测定
1.原理
当液体温度升高时,其蒸汽压随之增加,当液体的蒸汽压与大气压相同时,开始沸腾。液体在一个大气压时的沸腾温度即为该液体的沸点。
2.仪器
三口圆底烧瓶:有效容积为500mL。
试管:长190~200mm,距试管口15mm处有一直径为2mm的侧孔。
胶塞:外侧具有出气槽。
测量温度计:内标式单球温度计,精度为0.1℃,量程适合于所测试样的沸点温度。
辅助温度计:精度为1℃。
3.方法
见图8-2-33所示,将三口圆底烧瓶、试管及测量温度计以胶塞连接,测量温度计下端与试管液面相距20mm。将辅助温度计附于测量温度计上,使其水银球在测量温度计露出胶塞的水银柱中部。烧瓶中注入约为其体积的1/2的硫酸。

图8-2-33
1-三口圆底烧瓶 2-试管 3、4-胶塞 5-内标式单球温度计 6-辅助温度计 7-侧孔 8-温度计
量取适量试样,注入试管中,其液面略低于烧瓶中的硫酸的液面。加热,当温度上升到一定数值并在相当时间内保持不变时,此温度即为试样的沸点。记录下室温及气压。
4.气压对沸点影响的校正
根据测定时的室温及气压,按下式算出0℃时的气压:
p0=p1-△p
式中 p0——0℃时的气压,kPa;
p1——室温时的气压,kPa;
△p——室温时的气压换算至0℃时气压的校正值,kPa。
△p由表8-2-15查得,见表8-2-15。当0℃时的气压高于1013kPa时,自测得的温度值减去此校正值,反之则加。
表8-2-15 气压由室温换算至0℃时的校正值


5.测量温度计读数校正值的计算
若使用全浸式温度计进行测量,则应对温度计水银柱露出塞外部分进行校正。
温度计水银柱露出塞外部分的校正值按下式计算:
△t=0.00016h(t1-t2)
式中 △t——温度计水银柱露出塞外部分的校正值,℃;
h——温度计露出塞外部分的水银柱度数,℃;
t1——观测温度,℃;
t2——附着于1/2h处的辅助温度计温度,℃。
经第4条校正后的温度加上此校正值,即得到该试样的沸点温度。
(六)凝固点测定
1.仪器
装置见图8—2-34所示。

图8-2-34 凝固点测定装置图
1-温度计 2-干燥试管 3-套管 4-冷却浴
温度计:分度值为0.1℃的内标式单球温度计。
干燥试管:长150mm,内径20mm。
套管:长130mm,内径40mm。
冷却浴:盛有适宜的冷却液。
2.方法
称取15mL试样,置于干燥试管中。将温度计插入试管中,距管底10mm。然后将试管放入低于凝固点5~7℃的冷却浴中(不使温度计接触管壁),使试样冷却至低于凝固点3~5℃时,将试样迅速移入在上述冷却浴的套管中,用温度计搅动试样至试样凝固时,温度上升高达一定数值,并于该点保持一短暂时间不变,然后开始下降,取此短暂不变的温度作为试样的凝固点。
(七)结晶点测定
1.仪器
双层壁的玻璃试管(见图8-2-35):支管抽真空封闭。

图8-2-35 双层壁的玻璃试管
1-搅拌器 2-温度计 3-环形标线
圆筒型保温瓶。
水银温度计:-80~+60℃,分度1℃,2支。
搅拌器:铝或玻璃质。
试管架:供放双层壁试管用。
2.试剂
-20℃以上:食盐和碎冰。-20℃以下:乙醇干冰。
3.方法
①试样温度应调节至18~20℃,并混合均匀。
②将试样注入试管至标线处,并插入水银温度计,其位置处于试管中央与管底距离为15mm。
③容器中所贮冷剂液面高于试样液面30~40mm,使冷剂的温度自始至终比试样温度低15~17℃,并以每分钟60~200次的速度搅拌试样。估计达到结晶点前5℃时,从冷剂中取出试样,置于光源下仔细观察。如试样未呈晶体,将试管放入冷剂中,以后每隔1℃观察一次,每次观察时间不超过12s。
当试样开始呈现出肉眼所能见的晶体或呈凝胶状时,所示温度即为其结晶点。
(八)皂化值及酯含量测定
1.仪器
锥形瓶:250mL(磨口,无硼耐碱)。
回流冷凝器:磨口。
滴定管:50mL。
水浴。
移液管:50mL。
2.试剂
95%乙醇。0.5mol/L氢氧化钾乙醇溶液:称取28g氢氧化钾溶于100mL蒸馏水中,待完全溶解后,将其倒入900mL95%乙醇,摇匀即可。盐酸:0.5mol/L标准溶液。酚酞:1%乙醇溶液。
3.方法
称取0.5~1g试样,准确至0.0002g,置于锥形瓶中,加入50mL0.5mol/L的氢氧化钾乙醇溶液,然后装上回流冷凝管,在沸水浴上加热回流0.5~2h,用少量无二氧化碳蒸馏水(约10mL)冲洗冷凝管管壁,趁热取下皂化瓶,加2~4滴酚酞指示剂,以0.5mol/L的盐酸标准溶液滴定至终点,同时做一空白试验。
4.计算
增塑剂皂化值按下式计算:

增塑剂酯含量按下式计算:

式中 V1——试样消耗盐酸标准溶液的体积,mL;
V2——空白试验消耗盐酸标准溶液的体积,mL;
c——盐酸标准溶液的浓度,mol/L;
M——增塑剂的摩尔质量,g/mol;
n——酯的价数;
X——酸值换算为酯含量及皂化值的数(酸值小于1mgK()H/g可忽略不计);
m——试样的质量,g;
56.11——氢氧化钾相对分子质量。
平行测定两个结果的差数不应大于0.5%(酯含量),及2mg KOH/g(皂化值)。以平行测定两个结果的算术平均值为增塑剂的酯含量及皂化值。
(九)含水率测定-碘、二氧化硫法
1.仪器
仪器如图8-2-36所示。
(1)玻璃下口瓶,容积70~100mL,具有侧孔(磨口)及带玻璃截门的下口。
(2)半微量自动滴定管。
(3)铂电极。
(4)电磁搅拌器。
(5)微安计。
(6)干电池:15V。
(7)可变电阻器:2000Ω。
2.安装
见图8-2-36所示,将玻璃下口瓶用胶塞与滴定管、铂电极连接,侧孔与具有磨口的硅胶干燥管连接。测定时,试样溶液由侧管加入。用“死停”法确定终点时,将1.5V干电池、2000Ω的可变电阻器、微安计和铂电极如图连接。

图8-2-36
1-电磁搅拌器 2-玻璃下口瓶 3-侧管 4-硅胶管 5-微安计 6-可变电阻器 7-干电池 8-铂电极 9-半微量自动滴定管
3.试剂
无水甲醇:取5g表面光洁的镁条及0.5g碘,置于圆底烧瓶中,加约70mL甲醇。回流至金属镁全部转变成白色絮状的甲醇镁。再加约900mL的甲醇,继续回流30min,然后进行分馏。在64~65℃收集无水甲醇。
无水吡啶:将试剂吡啶加适量氢氧化钠,回流4h,再进行分馏,在114~116℃收集无水吡啶。
碘:将升华碘于硫酸干燥器内干燥48h以上。
碘-二氧化硫溶液:称取110g碘置于干燥的1000mL烧瓶中,加160mL无水吡啶,振摇,待碘全部溶解后,加300mL无水甲醇,在冰浴中冷却。然后通入经过硫酸干燥的二氧化硫,使增质量达72g,再加无水甲醇使体积为1000mL。放置暗处24h后可用。
水标准溶液:称取0.2~0.25g水,称准至0.0002g。用无水甲醇准确稀释至100mL。
4.终点确定
(1)目测法:取2mL无水甲醇,用碘-二氧化硫溶液滴定至溶液由浅黄色变为浅棕黄色。
(2)“死停”电位法:取2mL无水甲醇,用碘-二氧化硫溶液滴定,调整电路中电阻,使过量1滴溶液(约0.02mL)产生25~50μA的电流。滴定至指针偏转至某一位置,不再发生倒转现象即为终点。
5.方法
(1)碘-二氧化硫溶液的标定:
空白试验:取2mL无水甲醇在搅拌下用碘-二氧化硫溶液滴定至终点,不记录读数,再取5mL无水甲醇,继续滴定至终点,记录消耗的体积。
标定:取2mL无水甲醇在搅拌下用碘-二氧化硫溶液滴定至终点,不记录读数,再取5mL水标准溶液,继续滴定至终点,记录消耗的体积。
按下式计算碘-二氧化硫对水的滴定度T:
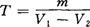
式中 V1——标定时碘-二氧化硫溶液消耗的体积,mL;
V2——空白试验时碘-二氧化硫溶液消耗的体积,mL;
m——5mL水标准溶液中含水之质量,g。
(2)含水率测定:取2mL无水甲醇,在搅拌下用碘-二氧化硫溶液滴定至终点,不记录读数,然后迅速加入5g(4.3mL)试样,继续滴定至终点。
6.计算
按下式计算试样的含水率:

式中 w——试样的含水率,%;
T——碘-二氧化硫溶液对水的滴定度,g/mL;
m——试样质量,g;
V1——碘-二氧化硫溶液消耗的体积,mL;
V2——试样的体积,mL;
d——试样的相对密度。
(十)酸值测定
1.对易溶于乙醇的增塑剂的酸值测定
(1)仪器:
微量滴定管:1~2mL,分度0.02mL;
磨口锥形瓶:100~125mL。
(2)试剂:0.2mol/L及0.5mol/L95%氢氧化钾乙醇标准溶液。0.2mol/L及0.5mol/L氢氧化钠标准溶液。碱性蓝6B乙醇溶液:称取碱性蓝6B1g(准确至0.01g),放入50mL煮沸的95%乙醇中,并在水浴上回流1h,冷却后过滤,必要时,将加热的澄清液用0.5mol/L氢氧化钠标准溶液或0.5mol/L盐酸溶液中和,直至加1~2滴碱溶液并使指示剂溶液从蓝色变成浅红色,冷却后又能恢复为蓝色。
酚酞:1%乙醇溶液。
苯酚红:将0.10g苯酚红指示剂溶于14.20mL0.2mol/L氢氧化钠溶液中,用水稀释至250mL。
(3)方法:取100mL乙醇,加2滴酚酞指示剂,用0.2mol/L或0.5mol/L氢氧化钾(或0.2mol/L或0.5mol/L氢氧化钠标准溶液)标准溶液中和至粉红色备用。
称取5~10g试样,准确至0.01g,置于具有磨口塞的锥形瓶中,然后分别以50mL已中和的乙醇置于两瓶中,其中不加试样乙醇作为终点比色标准。另一瓶需待测试样全溶后,以0.2mol/L或0.5mol/L氢氧化钾乙醇标准溶液(或0.2mol/L或0.5mol/L氢氧化钠标准溶液)滴定至与标准颜色相同(滴定需在30s内完成),保持15s不退色即为终点。
(4)计算:增塑剂酸值X(mgKOH/g)按下式计算:

式中 V——耗用氢氧化钾或氢氧化钠标准溶液的体积,mL;
c——氢氧化钾或氢氧化钠标准溶液的浓度,mol/L;
m——试样的质量,g;
56.11——氢氧化钾相对分子质量。
以平行测定两个结果的算术平均值为增塑剂的酸值。
2.对不易溶于乙醇的增塑剂的酸值测定
(1)仪器:
微量滴定管:1~2mL,分度0.02mL;
磨口锥形瓶:100~125mL。
(2)试剂:
无水乙醇。95%乙醇。石油醚:馏程90~120℃。0.2mol/L及0.5mol/L氢氧化钾乙醇标准溶液。溴甲酚酯:1%乙醇溶液。酚酞:1%乙醇溶液。石油醚-乙醇混合溶液:按体积1:1配制。
(3)方法:取100mL石油醚-乙醇混合溶液,加1mL溴甲酚酯指示液(或酚酞指示剂),用0.5mol/L氢氧化钾标准溶液中和至绿色。
称取5~10g试样,准确至0.01g,置于具有磨口塞的锥形瓶中,然后分别以50mL已中和的石油醚-乙醇混合溶液,待试样完全溶解后,以0.5mol/L氢氧化钾乙醇标准溶液滴定至与标准颜色相同(滴定需在30s内完成),保持15s不退色即为终点,同时用不加试样的石油醚-乙醇混合溶液作为终点比色标准。
(4)计算:
增塑剂酸值X(mgKOH/g)按下式计算:

式中 V——耗用氢氧化钾乙醇标准溶液的体积,mL;
c——氢氧化钾乙醇标准溶液的浓度,mol/L;
m——试样的质量,g;
56.11——氢氧化钾相对分子质量。
以平行测定两个结果的算术平均值为增塑剂的酸值。
(十一)碘值测定
该方法适用于环氧酯碘值的测定。
1.仪器
碘量瓶:250mL;
移液管:25mL;
量筒:10mL、15mL;
碱式(棕色)滴定管:50mL。
2.试剂和溶液
甲醇。溴化钠。三氯甲烷。四氯化碳。碘化钾:15%水溶液。0.05mol/L硫代硫酸钠标准溶液。0.5%淀粉指示液。0.1mol/L三溴化合物甲醇溶液:取76g无水溴化钠(在恒温箱中,保持130℃恒温3h),溶于1000mL甲醇中,加入5.1mL溴,贮存于棕色试剂瓶中,混匀,过一昼夜用。
3.方法
准确称取0.6~1.2g试样(视碘值大小而定),准确至0.0002g,移入250mL碘量瓶中,加入10mL三氯甲烷(或四氯化碳)溶解。用移液管准确加入25mL三溴化合物甲醇溶液,塞紧瓶塞,混匀。放置暗处静置20min(或振摇5min),然后加入15mL15%碘化钾溶液和75mL水,用0.05mol/L硫代硫酸钠标准溶液滴定至淡黄色,加入1~2mL0.5%的淀粉指示液,继续滴定至蓝色消失,同时做空白试验。
4.计算
增塑剂碘值X按下式计算:

式中 V1——滴定空白消耗硫代硫酸钠标准溶液的毫升数;
V2——滴定试样消耗硫代硫酸钠标准溶液的毫升数;
c——硫代硫酸钠标准溶液的浓度,mol/L;
m——试样的质量,g;
0.2538——碘的毫摩质量。
以平行测定两个结果的算术平均值为增塑剂的碘值。
(十二)环氧值测定
该方法适用于环氧增塑剂环氧值的测定。
1.盐酸-丙酮法
(1)仪器:
具塞磨口三角锥形瓶:250mL;
容量瓶:1000mL;
碱式滴定管:50mL,分度0.01mL。
(2)试剂和溶液:
盐酸、丙酮、0.01mol/L氢氧化钠标准溶液、0.15mol/L氢氧化钠标准溶液、95%乙醇、酚酞、甲酚红(邻位甲酚磺钛)、百里香酚蓝、盐酸-丙酮溶液:取盐酸1份,丙酮40份(以体积计)混合,密闭贮存于玻璃瓶中备用。
酚酞指示液:取酚酞1g溶于100mL乙醇。
混合指示液:
①0.1%甲酚红溶液:准确称取甲酚红100mg(准确至0.0002g),加入26mL0.01mol/L氢氧化钠标准溶液,溶解后加蒸馏水稀释至100mL。
②0.1%百里香酚蓝溶液:准确称取百里香酚蓝100mg(准确至0.0002g),加入22mL0.01mol/L氢氧化钠标准溶液,溶解后加蒸馏水稀释至100mL。
取0.1%甲酚红溶液10,加0.1%百里香酚蓝溶液30mL,混合均匀,以0.01mol/L氢氧化钠标准溶液及0.01mol/L盐酸标准溶液调至中性(混合指示剂约在pH9.8时变色)。
(3)方法:准确称取0.5~1.0g试样,准确至0.0002g,置于250mL具塞磨口三角锥形瓶中,准确加入盐酸-丙酮溶液20mL,塞紧瓶塞,混匀。放置暗处静置30min。加入混合指示液5滴,用0.15mol/L氢氧化钠标准溶液滴定至紫蓝色,同时做空白试验。
(4)计算:
增塑剂环氧值X按下式计算:

式中 V——空白试验消耗氢氧化钠标准溶液的毫升数;
V1——试样试验消耗氢氧化钠标准溶液的毫升数;
V2——试样中测定酸值消耗氢氧化钠标准溶液的毫升数;
c——氢氧化钠标准溶液的浓度,mol/L;
m——试样的质量,g;
ms——测定酸值时试样的质量,g;
0.032——氧的毫摩质量。
以平行测定两个结果的算术平均值为增塑剂的环氧值。
2.盐酸-吡啶法
(1)仪器:
具塞磨口三角锥形瓶:250mL;
碱式滴定管:50mL,分度0.01mL;
直形冷凝管:长40cm;
油浴锅。
(2)试剂和溶液:
盐酸、吡啶、丙酮、0.2mol/L氢氧化钠标准溶液。95%乙醇、酚酞、盐酸-吡啶溶液:取盐酸16mL加984mL吡啶,混匀后密闭贮存备用。酚酞指示液:取酚酞1g溶于100mL95%乙醇。
(3)方法:准确称取0.5~1.0g试样,准确至0.0002g,置于250mL具塞磨口三角锥形瓶中,准确加入盐酸-吡啶溶液20mL,装上回流冷凝器,置于油浴锅内,加入回流20min(油浴温度128℃),放冷后用15mL中性丙酮冲洗冷凝管,加入酚酞指示液4~5滴,用0.2mol/L氢氧化钠标准溶液滴定至粉红色,同时做空白试验。
(4)计算:增塑剂环氧值X按下式计算:

式中 V——空白试验消耗氢氧化钠标准溶液的毫升数;
V1——试样试验消耗氢氧化钠标准溶液的毫升数;
V2——试样中测定酸值消耗氢氧化钠标准溶液的毫升数;
c——氢氧化钠标准溶液的浓度,mol/L;
m——试样的质量,g;
ms——测定酸值时试样的质量,g;
0.032——氧的毫摩质量。
以平行测定两个结果的算术平均值为增塑剂的环氧值。
(十三)灰分测定
1.仪器
带盖的石英坩埚或瓷坩埚,容量30mL。
干燥器,内盛变色硅胶。
石棉板。
坩埚钳。
电炉。
高温炉或坩埚炉,附温度调节装置。
2.试剂和溶液
盐酸、硝酸铵。
3.方法
用盐酸溶液(1∶4)洗涤坩埚,并用蒸馏水洗净,然后放在高温炉或坩埚炉内,按测定试样的温度和时间条件预先加热处理,经冷却,称量,准确至0.0002g。准确称取10g试样,准确至0.01g于加热处理并称量后的坩埚中,置电炉上慢慢加热(防止起火),蒸发近干。然后,将坩埚移入600℃±30℃高温炉或坩埚炉内,灼烧1~1.5h至残留物灰化(难灰化的增塑剂可在残留物中加入1~2g硝酸铵)。残留物灰化后,将坩埚取出,稍冷约2min,移至干燥器内,冷却至室温(约30min),称量(准确至0.0002g)。再置于炉内重复灼烧0.5h,冷却,称量。如此操作直至连续两次称量的质量变化不超过0.0005g。
4.计算
增塑剂灰分质量分数X(%)按下式计算:

式中 m1——灼烧后灰分与坩埚的质量,g;
m2——坩埚质量,g;
m——试样质量,g。
(十四)砷测定(砷斑法)
1.仪器
定砷器,见图8-2-37所示。

图8-2-37 定砷器
1-广口瓶 2-胶塞 3-玻璃管 4-玻璃管上端管口 5-玻璃帽
(1)广口瓶:容积125mL。
(2)玻璃管:长180mm,上部直径6.5mm,管的末端有一直径约为2mm的孔。使用前装入乙酸铅棉花,高约60mm。
(3)玻璃管的上端管口:表面磨平,下面有四个耳钩,供固定玻璃帽用。
(4)玻璃帽:下面磨平,中央有孔与玻璃管相通,孔直径6.5mm上面有弯月型凹槽。
2.试剂
砷、盐酸、碘化钾、氯化亚锡、锌:无砷、溴化汞试纸。
3.方法
称取适量试样,溶于水,注入广口瓶中,稀释至70mL。加6mL盐酸,摇匀,加1g碘化钾及0.2mL40%氯化亚锡溶液,摇匀,放置10min。加2.5mg无砷金属锌,立即按图装好装置,于暗处在25~30℃放置1~1.5h。溴化汞试纸所呈棕黄色不得深于标准。标准是取规定数量的砷,与试样同时同样处理。
(十五)重金属测定
称取1.5g试样,加40mL水溶解,然后加1mL1mol/L乙酸及10mL新制备的饱和硫化氢水,摇匀,放置10min,所呈暗色不得深于标准(标准是取0.5g试样,溶于25mL水中,加入0.01mg的铅杂质标准溶液,稀释至40mL,与同体积试样溶液同时同样处理)。
(十六)运动粘度测定
1.品氏法
该方法适用于运动粘度在0.001~15Pa·s的液体增塑剂运动粘度的测定。当粘度大于15Pa·s时,应选用直径为5.0mm或6.0mm的品氏粘度计进行测定。
(1)仪器:
①品氏毛细管粘度计一组,每支具有3处扩张部分(见图8-2-38)。各支的毛细管内径分别为0.4mm、0.6mm、0.8mm、1.0mm、1.2mm、1.5mm、2.0mm、2.5mm、3.0mm、3.5mm、4.0mm、5.0mm、6.0mm,每支粘度计必须有粘度计常数。

图8-2-38 品氏毛细管粘度计
1-毛细管 2,3,5-扩张部分 4-管身 6-支管 a、b-标线
测定试样的运动粘度时,应根据试验的温度选择适当的粘度计,务使试样的流动时间能在300s±180s范围内,选用内径适当的毛细管粘度计。
②带有透明壁或装有观察孔的恒温器,高度不小于180mm,容积不小于2L,并附有设着自动搅拌装置和一种能够准确地调节温度的电热装置(最好同时采用温度调节器)。缺乏恒温器时,可以使用烧杯,其高度不小于170mm,容积不小于1L。
根据测定的条件,要在恒温器或其他容器中注入见表8-2-16所列举的一种液体。
表8-2-16

(2)试剂:橡胶溶剂油或石油醚;乙醚;乙醇(化学纯)。
(3)准备:
①测定增塑剂的粘度之前,必须将粘度计用橡胶溶剂油、轻汽油、石油醚或乙醚洗涤,如果粘度计有污垢,就用铬酸洗液、蒸馏水或乙醇等仔细地洗涤,然后放入烘箱中烘干或用通过棉花滤过的热空气吹干。
②使用品氏粘度计测定运动粘度时,在内径符合要求的清洁干燥毛细管粘度计内装入增塑剂。装入之前将橡皮管套在支管6上,并用手指堵住管身B的管口,同时倒置粘度计。然后将管身4插入装有试样的容器中,这时利用橡皮球、水流泵或其他真空泵将液体吸到标线b,同时注意不要使管身4、扩张部分2和3中的液体发生气泡或裂隙,当液面达到标线b时,就从容器提起粘度计并迅速恢复正常状态。同时将管身4的管端外壁所粘着的多余试样擦去,并从支管6取下橡皮管套在管身4上。
③将装有试样的粘度计浸入事先准备妥当的恒温器中,并用夹子将粘度计固定在支架上,在固定位置时品氏毛细管粘度计必须把扩张部分3浸入一半。温度计要用另一支夹子来固定,务使水银球的位置接近毛细管1中央点的水平面,最好使温度计上要测温的刻度位于恒温器的液面上20mm。
使用全浸式的温度计时,如果它的测温刻度露出在恒温器的液体上面,就依照下式计算水银柱露出部分的修正数△t,才能准确地量出液体的温度。
△t=kh(t2-t1)
式中 t1——测定粘度时的规定温度,℃;
t2——接近水银柱露出部分的空气温度,℃(用另一支温度计量出);
k——常数,水银温度计采用k=0.00016;
h——在液面上露出的水银柱高度(用温度计的度数表示)。
试验时取t1与△t的代数和,作为温度读数。
(4)方法:
①将粘度计调整为垂直状态,要利用铅垂线从两个相互垂直的方向取检查毛细管的垂直情况。将恒温器调整达到规定的温度,将装好试样的粘度计浸在恒温液体内,经过见表8-2-17规定的时间。
表8-2-17

②使用品氏毛细管粘度计测定运动粘度时,利用管身4所套着的橡皮管将试样吸入扩张部分2,使液面稍高于标线a,并且注意不要让毛细管和扩张部分2中的液体发生气泡或裂隙。
③此时观察试样在管身A中的流动情况。液面正好达到标线a时开动秒表,液面正好流动到标线b时,停住秒表。
试样的液面在扩张部分2(或扩张部分3)中流动时,注意恒温器中正在搅拌的液体要保持恒定温度,而且扩张部分中不应出现气泡。
④用秒表记下流动时间,应重复测定至少4次,其中各次流动时间与算术平均值的差数应符合下列要求:在-30℃以上测定粘度时,差数不应超过算术平均值的0.5%。然后取不少于3次的流动时间所得的算术平均值,作为增塑剂的平均流动时间。
(5)计算:在温度t时试样的运动粘度vt(10-3Pa·s)按下式计算:
vt=ctt
式中 c——粘度计常数,Pa;
τt——试样的平均流动时间,s。
测定试样的运动粘度时应在每一次试验温度进行两次测定,两次测定中,测定结果与算术平均值的差不得超过±10-2Pa。以测定结果的平均值作为试样的运动粘度
2.恩氏法
该方法适用于动力粘度在10Pa·s以下的液体增塑剂动力粘度的测定。
恩氏粘度是试样在某温度,从恩氏粘度计流出200mL所需的时间与蒸馏水在20℃流出相同体积所需的时间(s)(即粘度计的水值)之比。
(1)仪器:
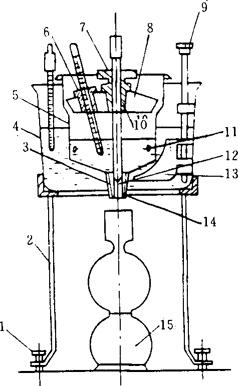
图8-2-39 恩氏粘度计
1-调整螺钉 2-铁三角架 3-铂制小管 4-外容器 5-内容器 6-插温度计孔 7-木塞孔 8-黄铜盖 9-搅拌手柄 10-木塞 11-小尖钉 12-流出孔 13-搅拌器 14-黄铜小管 15-接收瓶,20℃时为200mL
①恩氏粘度计(见图8-2-39)。
②恩氏粘度计用的温度计。
③恩氏粘度计用的接受瓶。
④电加热装置。
⑤吸量管:容积5mL。
⑥秒表:分度为0.2s,经过校正。
(2)试剂:
石油醚、乙醚或乙醇(化学纯)。
(3)方法:
①用石油醚、乙醚或乙醇和蒸馏水将粘度计内容器洗涤并用空气吹干,用铬酸洗液、水和蒸馏水将接收瓶洗涤,将新蒸馏的蒸馏水(20℃)装入内容器和外容器。将接收瓶再置于流出管下。
②水值的测定,将内容器水面调至三个尖钉的尖端刚露出水面为止,将外容器的水调至扩大部分为止。
很好搅拌内容器和外容器中的水,首先将插有温度计的盖围绕木塞旋转,然后用安在外容器上的叶轮式搅拌器搅拌,确保两容器中的水温度等于20℃(在5min内的温度差数不大于±0.2℃)。当内容器调整为水平线(三个尖钉端刚好在水面露出)时迅速略提木塞(自动保持稍微提起的状态),同时开动秒表,此时观察水从内容器流出的情况,到凹液面下边缘达到接收瓶的200mL刻线时立即停止秒表。
连续测定4次,每次测定结果与平均值之差不得大于1,取其算术平均值。
标准粘度计的水值为51s±1s。恩氏粘度计的水值每4个月至少检查一次。
③将恒温的试样注入内容器(注意不要产生气泡),注入的液面,必须稍离于尖钉尖端。以测水值法测定试样流速。
(4)计算:在温度t时试样的恩氏粘度Et(以度为单位)按下式计算:

式中 τt——在试验温度t时从粘度计中流出200mL的时间,s;
k20——粘度计水值,s。
两次平行测定时间的差数不应超过下列数值:
两次时间/s 允许误差/s
250以下 1
251~500 3
501~1000 5
大于1000 10
取平行测定两个结果的算术平均值作为试验的恩氏粘度。
(十七)加热减量测定
该方法是在规定温度和时间的条件下,对加热后增塑剂所损失的质量分数进行测定。
1.仪器
电热恒温干燥箱:内层(250~500mm)×(240~500mm),恒温波动范围±2℃,内装玻璃门。
称量瓶:直径40mm,高25mm。
干燥器。
圆形石棉板:直径200mm,厚2.3mm。
2.方法
将电热恒温干燥箱调节至125℃±2℃,在已恒量的两个带盖称量瓶中,分别称入6~8g(准确至0.0002g)试样。将盛有试样的称量瓶置于石棉板的适当位置,并一起放入电热恒温干燥箱中。试样液面应距温度计水银球底部10~20mm,称量瓶的对称轴线与温度计的距离不超过70mm,自温度回升到测试温度起保持2h,然后将称量瓶移入干燥器内,称量。
3.计算
增塑剂的加热减量X(%)按下式计算:

式中 m1——加热前试样与称量瓶质量,g;
m2——加热后试样与称量瓶质量,g;
m——试样原质量,g。
平行测定2个结果的差数不大于0.05%,以平行测定两个结果的算术平均值作为试样的加热减量。
(十八)热稳定性试验
该方法是在一定的温度下对增塑剂加热后色泽和酸值的变化进行测定。
1.仪器
硬质试管:内径38mm,长200mm。
加热器:双孔铝制加热器(见图8-2-40),电源220V,控制电压70V,电功率500W控制温度180℃±2℃。
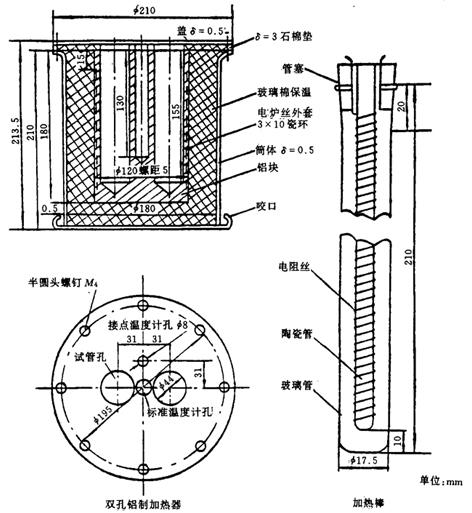
图8-2-40 双孔铝制加热器
加热棒:电源220V,控制电压40V,电功率100W。
调压变压器:2台,1kVA。
自动恒温控制器。
水银接点温度计:150~210℃,分度为1℃。
温度计:150~210℃,分度为1℃,2支。
锥形瓶:500mL,2个。
2.方法
(1)取试样70mL或105mL,置于硬质试管中,盖一铝片,铝片上有两个孔,分别为温度计和加热棒的插入口。温度计水银球处于液面下4cm处。当铝制加热器恒温到180℃±2℃时,将试管放入加热器的孔中,同时迅速将加热棒放入试管中,并调节加热棒的变压器,使试样在8~10min内温度上升到180℃±2℃,并保持到取出为止。从试管放入加热器算起,经30min取出,将试样倒入500mL锥形瓶中,在5min内冷却至室温,测定色泽和酸值,每次同时测定两个试样。
(2)酸值测定方法与外观酸值测定(铂-钴比色法)相同,见本节(二)。
(3)酸值的测定见本节(十一)之1。