用农杆菌共培养或用Ti质粒转化原生质体
出处:按学科分类—生物科学 农业出版社《植物细胞培养手册》第349页(14474字)
本世纪初孟德尔法则作为遗传基础时,植物育种家就用有性杂交和选择方法改良作物。近30年来,分子生物学研究为植物细胞的遗传操作提供可能性。新技术发展一旦和传统植物育种技术相结合,就可成为有用工具。现代分子生物学主要研究基因结构和表达的调控。DNA重组技术使得分离基因成为可能。近年来原生质体分离方法得到发展,原生质体能人工培养和再生植株。同时,认识到农杆菌可作为遗传物质导入植物细胞的非常有效的媒介体。本章主要叙述依靠肿瘤农杆菌把外源DNA导入植物细胞的自然能力和单细胞(原生质体)转化体系的发展。
(一)天然基因转移体系
细胞核和细胞质基因被确认为用来转移入植物细胞的因素。实际上,用重组DNA有利于转移任何基因,包括微生物,进入植物细胞。但是要达到这一点必须满足许多条件:首先是往复性基因载体,它必须含有能在细菌里同样在植物细胞里复制和选择的DNA顺序,并有可能加以操作。为了在细胞分裂和有性传代期间,导入的基因的稳定保持。带有DNA序列的载体比自发复制的载体更有利于保证外源DNA与受体基因组相整合。DNA重组技术能构成携带染色体着丝粒和自发复制(AR)顺序的极微染色体。这些AR顺序,以有规律距离存在于真核染色体的DNA中。在酵母上,极微染色体和AR顺序已成功地插入质粒。植物病毒也可用作载体,表现自发复制。植物病毒以RNA为遗传物质,可经cDNA制成载体,遗憾的是用这些病毒进展很慢。目前着重于用含DNA病毒如花椰菜嵌合病毒(CaMV),把外源DNA插入CaMV基因组,通过感染敏感型寄主植物把DNA导入细胞。尚未发现植物病毒能把它们的遗传信息导入寄主基因组。采用导入的DNA片段包埋法已有进展。但是,植物病原体肿瘤农秆菌和发根农杆菌能把它们的DNA的特定部分导入植物细胞。这些农杆菌的能以诱导肿瘤(Ti质粒)和毛根病(Ri质粒)的大质粒能把它们的致病能力传给双子叶植物(寄主)。由于Ti或Ri质粒的特定部分的导入和整合植物染色体的结果,产生这些病征。细菌并不穿过被转化的细胞,它们牢固地附着在细胞的受伤侵染区域的细胞壁上,而转移DNA。本章讨论Ti质粒及其用作载体的一般概念。
有了好的载体系统,基因转移的第二个先提条件是有价值基因的分离和特征化。虽有相当多基因可从病毒和细菌染色体直接分离取得,但要从真核生物染色体分离特定基因是相当困难的。虽然有些基因复本或复基因系,但另一些并非如此。许多农业生产上重要复杂性状,如产量、对生物和非生物因素抗性,是多基因性状,既不很清楚,在生化上尚未经鉴定。正在研究分离基因的几种很微妙方法。
有了适宜的植物载体和适当DNA,已有方法把这种DNA导入植物细胞,并得以表达。为了使这类基因表达成功,要造成载体结构,使有价值基因受控于质粒基因的调控顺序(促进子区域)。这能作为这种载体的标志基因。
1.Ti质粒 Ti质粒存在于土壤细菌-肿瘤农杆菌里,是自然存在的一种转化体系,能满足上述条件,可用作载体。植物细胞繁殖过程中,肿瘤农杆菌引起冠瘿肿瘤病。自发转化的冠瘿瘤组织不同未转化组织,在不含激素的合成培养基上能生长。已知Ti质粒带有形成肿瘤,在瘿瘤细胞内合成冠瘿碱、细菌异化冠瘿碱和接合作用的遗传信息。Ti质粒视肿瘤产生的冠瘿类型而分成章鱼碱、蓝曙红或亮氨酸冠瘿碱Ti质粒。
冠瘿瘤是自然界通过重组DNA进行遗传操作的明显事例(图16-4)。农杆菌Ti质粒的一部分-Ti区域导入植物细胞的机制尚不清楚。细菌DNA与植物细胞核染色体DNA重组,这种外源DNA片段叫T-DNA,而成为植物基因的一部分。T-DNA可亚分为左(TL-)和右(TR-)DNA。后者能不存在于肿瘤中。T-DNA大小在8-16Mdal间。T-DNA转录成各种带有聚A末端的RNA分子,这是真核植物mRNA的典型状态。有必要更多地了解Ti质粒在植物细胞内对T-DNA转导、整合和表达所需基因和顺序。本章将详细讨论有关章鱼碱型质粒的表型特性的已有认识(图16-5)。

图16-4 肿瘤型毒性Agrobacterium fumefaciens侵染植物细胞发生的事项代表概图

图16-5 肿瘤农杆菌侵入植物细胞的发生过程
图示肿瘤型Ti质粒上最重要标志,肿瘤Ti质粒可分成二半,诱导产生肿瘤只要其中之一。这一半带有T-区域和vir区域。另一半带有章鱼碱(occ)、精氨酸(arc)和结合基因(tra)、除开噬菌体(ape)、不亲和(inc)和复制起点(ori)的降解或异化。T-区域带有生长素(aux)和细胞分裂素(cyt)活性基因,和在肿瘤细胞内合成章鱼碱基因。它的边际有24对碱基的直线诸重复,叫边际或连接顺序。vir-区域带有包括未知代谢的诸基因,由此T-区域成为TDNA转入和整合植物细胞.
分子遗传研究表明,约为质粒的一半不携带包含在致瘤内的任何基因。它主要具有利用章鱼碱和精氨酸代谢功能。结合和排斥菌体的基因也位于Ti质粒的代谢或降解区域。致瘤基因位于另一半基因组上的二个区域,叫毒性区(vir区域)和T-区。
T-区携带致瘤基因,在植物细胞内它的功能部分来自T-DNA。T-区诱变处理指出,它带有许多位点,以一些特定碱基顺序或重复顺序为其边际顺序。合成章鱼碱的基因在T-DNA上至少已鉴定出二个位点,控制着植物细胞生长和分化。用落地生根属或烟草植株突变体试验,当一个位点失活,从较小的瘤上长芽(芽突变体);另一个位点失活,则导致生根(根突变体)。二者的突变体的混合体的侵染,诱导正常肿瘤发生。当芽突变体加生长素或根突变体加细胞分裂素,产生同样结果。这种观察指出芽突变体受类生长素活性位点的影响(生长素位点),而根突变体则受细胞分裂素的影响(细胞分裂素位点),二者综合导致产生无组织肿瘤。这些位点可能直接担负着肿瘤组织的自主生长。研究这些T-DNA位点的功能和在植物细胞内它们的标的产物,可能控制植物细胞再生的分子过程提供线索。
Ti质粒的毒性区和T-区距离很近,T-区突变引起肿瘤变态,与此相反,毒性区突变可不形成或减少形成肿瘤(图16-6)。农杆菌侵染后产生的转化植物细胞内,未曾测得Ti质粒的毒性区,这个区的基因突变,能为农杆菌的反向品系所补偿,可见细菌的毒性的表达,受vir基因控制。其中有些担负着改造Ti质粒DNA的结构,成为出现在转化细胞内的T-DNA,通过识别T-区边缘顺序来完成。其它vir基因在建立Ti质粒DNA转入植物细胞的机制有重要性。

图16-6 基于T-区域和vir区域功能分开的双重载体策略
每种位于不同质粒上单是T-区域质粒(B)和vir区域质粒(A),不传给其寄主肿瘤发生。当具有亲和的质粒A和B在一个细胞内存在时,农杆菌具有一种诱导肿瘤的正常能力。这些质粒的复制以repl和rep2表示之。
2.Ti质粒作为植物基因载体用来导入植物基因组的基因,首先必须插入Ti质粒的T-区。Hoekema等(1983)构成二种亲和质粒,一种包含T-区,另一种包含章鱼碱Ti质粒毒性区。用这类质粒取得的结果,汇综如图16-6,表明一种双重载体策略。只携带T-区或毒性区的一种质粒农杆菌不能诱导产生肿瘤。当在一个细胞里具有两种质粒时,产生了诱导形成正常肿瘤能力和在肿瘤里测出章鱼碱的农杆菌。由此可知,T-区和毒性区可以机械地分开,而不失其致瘤作用。
带有遗传上操作的T-区的载体,可直接导入已具有毒性-质粒的农杆菌里。这种农杆菌可用于感染和转化植物细胞。但是,仍需外源DNA与植物基因组整合时,不带有T-区。已知致瘤基因缺失并不消失TDNA转入植物细胞和与之相整合的能力。这就指出带有只具有转移所需的T-区,如边缘顺序的构建,在有vir质粒存在下,可以转移。
很可能除这些边缘顺序外(约24个碱基),以及转入植物细胞整合的有用基因外,不需更多的DNA。由此能构成一种基因载体,在有vir区存在下,能把T-DNA区域转入植物细胞。图16-7表示只带有边缘顺序和冠瘿碱合成的标志基因的基因载体。此外,存在细菌操作的可选择标志(Em和Ap)和插入外源顺序的限制性酶切位点。致瘤基因不存在时,并不限制这种质粒的转化能力,并不破坏植物激素平衡和不形成肿瘤。不利的是不能根据植物激素独立性选择转化细胞。因此,要引入格外选择标志。用这类改建的载体,可任意地把基因导入植株。当植株受其感染后,伤口部位的一些细胞将被转导。但是由于不形成肿瘤,必须采用转化的细胞能在组织培养过程中生长,并能再生由全部转化细胞组成的植株的体系。
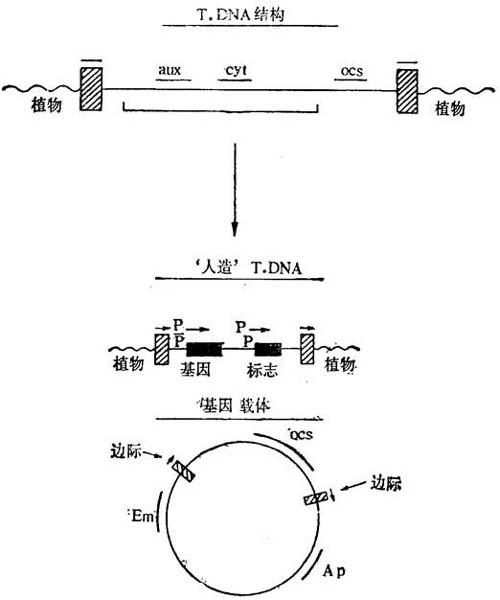
图16-7 把导入植物细胞的基因与在含有T-区域边际顺序(斜线区域)(基因载体)
诸片段间的选择或鉴定转化细胞的标志基因构成人工T-DNA存在于正常T-DNA上的一个基因,为其转入和整合植物染色体所不需。Aux和Cyt表示生长素和细胞分裂素位点。植物细胞内表达的促进子顺序以Pp表示。Ocs表示用作标志基因的章鱼碱合成酶,Em表示红霉素抗性,Ap氨 青霉素抗性。
青霉素抗性。
已有产生这类体系的二种方法。第一种方法是依靠农杆菌天然转化能力,即共培养程序。第二种方法是要避免农杆菌和双子叶植物之间的自发相互影响,称DNA转化方法。关于以卡那霉素抗性作为T-DNA在转化细胞中的鉴别标志,以代替致瘤标志的改建的Ti质粒。
(二)程序
1.植物原生质体的分离 植物细胞人工培养转化程序的发展,需要建立高植板率的原生质体良好体系,即有活力、细胞分裂以及生长的原生质体,即使转化频率低,也能有机会选择转化体。由于这种转化程序必将引起有些细胞死亡。我们多年来采用了良好规定的原生质体体系,证明适用于各种目的,变动培养基的荷尔蒙水平,也能再生植株。
无菌培养烟草变种Petit Havana SR1的芽,培养在T培养基,每隔4周继代。从3周龄芽取叶分离原生质体。除去主脉,其叶身下表面向下放在无菌培养皿中。加酶溶液,注意使全叶完全淹没。室温下暗培约18h。进一步培养在K3培养基+NAA0.54μM,KIN0.93μM。用NaOH调整到pH5.6,用前高温蒸气消毒。分离原生质体用酶为纤维素酶Onozuka R-10每15ml0.2g,离析酶Onozyka R-10,每15ml0.07g。通常1.5g叶片加过滤消毒的酶溶液15ml。原生质体产量为5-8百万。培养18h后,用手轻振动培养皿,以使从基质释放原生质体。然后把原生质体悬浮体经400和100μm金属滤网过滤,除去大碎片。随之洗原生质体,放入K3培养基,离心600rpm、纯化。重复两次。K1培养基含0.4M蔗糖,由此原生质体上浮。经轻度振荡后,原生质体在表面形成一层,使得能有效地与残屑分开,最后除尽培养基,其中含有残余化学试剂和酶。可用15cm长无菌玻璃毛细管连接蠕动泵插入原生质体层,然后用吸引法将原生质体层以下的培养液和残余物。计数原生质体,以适当密度悬浮在K3培养液中。全过程在超净台上完成。
2.通过共培养转化植物细胞 共培养程序包括肿瘤诱导,即在人工培养条件下,肿瘤农杆菌的天然感染过程(图16-8)。原生质体再生新细胞壁后,用毒性农杆菌与原生质体短期共培养。这样的原生质体的发育,好比受伤植物茎从分化的细胞产生条件性细胞。原生质体按图96方案处理。

图16-8 烟草原生质体与肿瘤农杆菌共培养和选择转化愈伤组织和芽的步骤概图
烟草变种Petit Havana SR1分离的原生质体,以密度104-105原生质体/ml悬浮在K3培养液中,含低生长调节剂:0.54μm NAA,0.93μmKIN。将装有10ml原生质体悬浮体的培养皿,暗培在26℃,24h,然后移入20001x48h。第3天即开始细胞分裂前,形成新细胞壁时(用荧光增白剂检测),将细胞与农杆菌以400∶1混合(细菌/原生质体比值)。共培养32h后,此时原生质体与细菌聚合,重复洗,除去游离细菌。进一步把植物细胞培养在加荷尔蒙的K3培养基中。加Carbenicillin 250μg/ml或cephotakime 100μg/ml以杀死残余细菌。3-4周后,将小愈伤组织植板在1∶10稀释的无荷尔蒙琼脂培养基上。连续选择压下,只有转化细胞组成的愈伤组织得以生存;由自发取得荷尔蒙自养细胞组成的驯化组织,在SR1原生质体中未曾产生。
当将毒性农杆菌加入新分离的或1天龄原生质体时,未观察到细菌的侵染和转化。3天龄原生质体新形成的细胞壁是农杆菌转化的重要条件。采用细胞壁形成抑制剂:香豆素、秋水仙素,2,6-二氯苯腈试验证实之。发现EDTA但不是EGTA(只与Ca2+相结合的试剂)能抑制侵染和转化。由于在施用这些螯合剂前,已形成了细胞壁,结果提出某些二价正离子如Mg2+而不是Ca2+,可能影响细菌与细胞相结合。诚如其它细菌-植物互作,外源凝集素可能起着结合过程中的重要作用。它的活性有赖于二价正离子的存在。从3天龄原生质体和原生质体生长的培养基提取的细胞壁材料,羧脯氨酸量增高。如果外源凝集素与糖类互作,在细菌侵染中起着作用,则在双子叶植物细胞壁的聚半乳糖醛酸与农杆菌壁的脂多糖(LPS)间起着桥梁作用。由于农杆菌寄主范围常广,任何化合物对有效侵染的有效控制,都不是专化的,无论植物种或农杆菌种。但是,可能有在农杆菌与双子叶植物间发生有效侵染中有特殊功能。就此观点,值得注意E.coli的LPS并不抑制肿瘤农杆菌的肿瘤诱导,和E.coli并不侵染3天龄原生质体。再度指出人工培养下观察到的农杆菌侵染,基本上不是非专化现象。与农杆菌有关的细菌,如Rhizobium trifolii能侵染再生细胞壁中的原生质体。证实了R.melilotii含有Ti质粒侵染的植物细胞,诱导产生肿瘤。但R.meliloti无此功能或侵染具有再生细胞壁的原生质体。经改造Ti质粒的农杆菌变为无毒性的,仍有侵染原生质体的能力。它们的LPS也能抑制由于毒性农杆菌诱导产生肿瘤。为此很可能农杆菌染色体上的基因决定着细菌侵染。
当用肿瘤农杆菌与16天的正在分裂中的原生质体发育成的正常烟草细胞集团共培养时,观察到侵染,但未见转化。在这种情况下,或是Ti质粒(或T-DNA)转移机制未能完全产生功能,或是质粒DNA转移了,但培养的烟草细胞是非“感受性”或“条件性”的,适应于稳定转化。从植株上肿瘤诱导得到相似结果,可作出结论:伤口细胞在发生分裂前是“条件性”的,此后并未发生转化。是否适合于所有植物种尚待研究。
冠瘿肿瘤与正常细胞不同,不为肿瘤农杆菌所侵染。曾观察到从烟草冠瘿肿瘤细胞分离的,或从含有T-DNA转化植株的叶的3天龄原生质体,为肿瘤农杆菌所结合。当肿瘤原生质体分裂和再生细胞团时,这种能力明显再次损失。指出可能存在于细胞壁内的为侵染所需的成分,是在原生质体时期合成的,但在细胞时期改变了,掩盖了或甚至损失了。
采用此法取得转化组织频率为开始时原生质体的0.1-1%。基于出现肿瘤标志、荷尔蒙自给和瘿瘤碱合成特征,指出这些标志集团存在于各种不同分离的肿瘤中。如表16-5所示。用相同菌族诱导植物产生的冠瘿肿瘤并未发现这种状态。Van Slogteren等(1983)假定原生质体共培养产生的转化体的T-DNA结构和组织是同质的,而活体植株上的冠瘿肿瘤则不同,予以解释。人工培养肿瘤,由肿瘤细胞混合群体所组成,如果有肿瘤标志存在,其分离由于互补作用而不能发现。采用原生质体分离,从互培养产生的亚克隆转化体系,也证实了离体培养肿瘤的同质性。不经选择可取得表型相同的亚克隆,带有与原来转化体相等的T-DNA结构。
表16-5 烟草原生质体与不同族的肿瘤农杆菌共培养后取得的转化体中的表型变异

a.Aut,荷尔蒙自养;Ocs,章鱼碱合成;Nos,蓝曙红合成
b.每种细菌约筛选200不同转化体。
通过共培养产生的章鱼碱转化体的T-DNA结构与植株上瘤形成的愈伤组织的T-DNA无显着差异。一般只含有相对固定大小的TL-DNA。观察到的表型变异,可能由于正常TL-DNA的偏差,或T-DNA基因的部分表达。例如,有些章鱼碱负转化体确含有合成章鱼碱的T-DNA基因,虽然不能测出RNA转录。这类情况下,章鱼碱不存在,或由于基因表达的控制。Van Slogteren等(1983)用胞嘧啶衍生物8-氮胞苷加入培养基,可能恢复章鱼碱合成,支持了这些观察结果。已知胞嘧啶能以甲基化,结果使DNA不能表达。8-氮胞苷不能甲基化。如果掺入DNA,可能在DNA中得到表达。这种现象可能说明某些转化的组织中不存在章鱼碱。
共培养的另一有意义状态,经常产生具有生芽能力高的章鱼碱型转化体。植株上野生型章鱼碱肿瘤,或从其产生的愈伤组织,未见芽的再生。在这类芽组织中,含有缺少正常Ti-DNA左侧部分的Ti-DNA,这些缺失至少部分影响着生长素位点,这可很好说明章鱼碱型处理的自发生芽能力。其特征之一是不生根。这种性质可能由于仍然存着的细胞分裂素位点的活性所致。
共培养方法已广泛用于引导不同来源基因进入若干植物种。大多数试验曾用所谓双臂Ti质粒,从此曾发生致瘤基因突变或缺失。带有这类族的农杆菌能完成DNA转移,但转化细胞不能在无荷尔蒙上生长。为此曾提出改变的选择程序,也通过Ti质粒导入新霉素磷酸转移酶(NPT-Ⅱ)基因,由此产生对抗生素卡那霉素或G-418的抗性。采用双臂Ti质粒的优点,在于从转化细胞能再生正常完整植株。把NPT-Ⅱ基因导入烟草细胞,或矮牵牛细胞,氯霉素乙酰转移酶(CAT)基因导入烟草细胞是其实例。这些基因都来自细菌。真核植物基因也曾转移。Murai等(1983)从豌豆种子蛋白质芸苔素转入烟草细胞,并能表达。Broglie等(1984)能以转移编码小亚单位核糖-1,5-2磷酸羧化酶Rubisco给矮牵牛细胞。细菌基因在植株内表达需要有Ti专化促进子嵌合基因,含有蓝曙红合成酶的5′促进子顺序以及采用细菌抗生素抗性基因编码区域。芸苔素和Rubisco小亚单位植物基因的表达,是在其自身促进子的转录控制下完成的。Rubisco小亚单位基因的表达是光依赖的。
3.植物原生质体通过Ti质粒转化DNA转化实验所用受体材料可用细菌到动、植物真核细胞。每个系统要有它本身的特殊条件以吸收DNA,但在若干方法中,但某些基本点是相似的。DNA可由四种途径供给受体细胞:(1)纯DNA,(2)磷酸钙-DNA共沉淀物,(3)DNA包埋在脂质体内,和(4)DNA直接微注射。热冲击、高pHl或加聚乙二醇(PEG),聚-L-氨酸(PLD)或聚乙烯醇(PVA)处理可增高DNA吸收。CaCl2处理能使受体细胞“感受”细菌。为了植物和酵母转化,必需用酶除去细胞壁以产生原生质体。不同生物体DNA转化的许多程序,典型地采用CaCl2和PEG组合处理。
磷酸钙-DNA互沉淀法的基本原则是在DNA存在下形成细小磷酸钙沉淀物。然后把单层细胞与DNA磷酸钙沉淀物共培养,而后培养在高浓度Ca2+培养基中。
还有微注射法。一般用拉丝机制成微吸管。采用显微操作器掌握吸管导入细胞;借助注射器压力将注射溶液导入细胞。这种方法最初用于大细胞如卵原细胞,目前也用于哺乳动物组织培养细胞,经固定化成为单层后施用。转化频率必需高,以克服能以操作的细胞数相对较少的状况。
(1)Ti质粒分离 把章鱼碱型肿瘤农杆菌族LBA4001分条接种在固化培养基上,以取得单克隆。采用TY培养基琼脂固化(tryptone 5g/l,酵母浸出液3g/l,琼脂18g/l)。取一个克隆悬浮在10ml TY培养液中,装入100ml欧氏三角瓶,29℃培养,连续振荡24h。然后把培养体加入3升欧氏三角瓶,装1L TY培养液,在相同条件下培养18h,一般用2L培养体,以分离质粒。培养体装入500ml吊桶,离心7000rpm20min。除去上清液,把细菌片状沉积物悬浮在1L T-E缓冲液中(Tris 50mM,EDTA20mM,用HCl调整pH到8.0)。悬浮物收集在玻璃烧杯中,用蛋白酶和1%(w/v)SDS溶菌30min,37℃。蛋白酶溶液:38ml T-E缓冲液中加240mg,预培养在37℃1.5h,以破坏任何污染的DNase。溶解后,当溶菌物完全清晰后,滴加3N NaOH使pH上升到12.2,恒常轻度搅动。继续培养10min。在这些条件下,只有染色体DNA将变性,而不是质粒DNA。以后滴加2M Tris pH7.0,使pH降到约8.6,把悬浮体加入1M NaCl。4℃培养4小时内,变性DNA将沉淀,经4℃离心7000rpm10分钟除去之。收集上清液加50%(w/v)PEG6000溶液,最后浓度为10%(w/v)。4℃培养过夜,使DNA沉淀,用4℃离心7000rpm10分钟取得片状沉积物,溶于二份25mlT-E缓冲液。取25gCsCl溶于每份DNA溶液,加1875ml溴化乙锭(EtBr 10mg/ml)。在装有荧光实验室制备这种溶液,以避免在紫外光影响下由于EtBr存在而DNA破坏。用异质同晶管(容积约30ml)装在60Ti转动机上进行浮力密度梯陡离心15℃32000rpm60h。离心后,在紫外光下可见二条带。收集最低的带由超旋Ti质粒组成,用20×SSC(1×SSC是15MNaCl,0.015MNatri-柠檬酸饱和的异构戌基醇)提取5次,以除去EtBr。这种DNA溶液放在4℃下分馏过夜,逆对3%(w/v)NaCl,溶于T-E缓冲液,用3%NaCl饱和的苯酚提取,再用氯仿:异构戊基醇(50:1)提取。重复苯酚和氯仿循环一次;加2体积冷96%乙醇-20℃过夜,从水相沉淀DNA。放在30ml Corex管内离心10000rpm4℃20min后,用70%乙醇洗DNA片状沉积物,再用96%乙醇洗,真空中干燥,溶于400μl 1mM Tris加0.4mM EDTA,pH7.00用于DNA转化试验前,用荧光计检测DNA量,随着限制性核酸内切酶解离,用琼脂糖凝胶电泳鉴定质粒的完整性。至少在目前,在无菌带塞试管内,加几滴氯仿,以消毒DNA溶液。DNA溶液的无菌性质,取部分DNA溶液用于DNA转化试验,取几滴余下的溶液植板在TY培养基上,放入培养皿,培养在29℃几天,加以核查。
(2)DNA和原生质体培养 DNA转化培养基叫F培养基,成分如下:NaCl 140mM,KC15mM,Na2HPO40.75mM,葡萄糖5mM,和CaCl2·2H2O125mM。制成100ml F培养基。调整pH到7.0。此时形成磷酸钙细粒沉淀。将其除去。由于它具有吸取DNA的作用,像PEG和高Ca2+浓度一样。固体PEG溶于F培养液中,最后浓度为40%(w/v)。高压蒸气消毒后,pH降到5.5,与原生质体悬浮体的K3培养基的pH相等。
取1ml原生质体悬浮物含原生质体5×105加入12ml Kimax离心管,与0.5ml 40%(w/v)PEG6000溶于F培养基相混合,继之加10μgTi质粒DNA(通常用25-50μl)和50μg牛胸腺DNA(50μl)。小牛胸腺DNA用作携带者DNA,从1mg/ml小牛胸腺DNA溶液中取得。用前用几滴氯仿消毒溶液。加入各种成分后,产生成团原生质体。培养基中转化诱导剂的最后浓度为13.3%(w/v)PEG,和约40mMCa2+。培养在26℃ 20001x30min,偶加振荡,使培养基与原生质体团接触良好,后者趋于浮于表面。全部程序如图16-9。

图16-9 原生质体转化程序图
(3)后培养和培养 培养后30min后,用2ml F培养基小心稀释培养基,PEG浓度降到5.7%,Ca2+浓度增至89mM。悬浮体静置于室温下(约21℃)5min。第二次加2mlF培养基,随之静置5min。这种处理重复3次,直到加了10ml培养基。这样PEG浓度逐级下降,而Ca2+则逐级上升。渐次加F培养基极为重要。后培养之末,PEG浓度约为1.6%(w/v),Ca2+约为115mM。取悬浮离心600rpm。由于降低蔗糖浓度,原生质体团和游离原生质体将沉降,用蠕动泵经玻璃毛细管虹吸易于除去上清液。原生质体悬浮在10mlK3培养液(0.4M蔗糖)中,含有250μg/ml carbenicillin,附加上述植物荷尔蒙。此时在DNA转化处理期间形成的大块细胞团,破碎成较小团块和许多游离原生质体。由于剩余PEG浓度已不足以影响原生质体成活率和培养,不再需要洗以除去PEG。再者,每次离心要损失原生质体至少10%。从K3培养液中取出原生质体,应立即放入培养皿。光学显微镜观察,通常转化后细胞生存率约50%。在此条件下,未见原生质体融合。如果发生,估计低于5%。在原生质体培养最初24h,暗培,26℃。
(4)生长和选择 原生质体培养基确实含有植物荷尔蒙,但其水平较愈伤组织培养基的为低。正常SR1烟草原生质体能生长在所用的培养基上,虽未经转化,仍需在培养早期有必要加植物荷尔蒙。当它们培养在无荷尔蒙培养基时,所用荷尔蒙浓度容许迅即选择非转化细胞。在人工培养DNA转化程序中,烟草细胞保持在较低水平荷尔蒙培养基上较长一个月,与通常的共培养程序相比。
暗培后,带有原生质体培养皿转入20001x每天12h,培养在26℃。2周后,取一半(约5ml)移入新培养皿,每皿加新鲜K3培养基5ml。2周后,活跃分裂中细胞悬浮体,用K3培养基以1∶20稀释,仍含荷尔蒙,但蔗糖降到0.3M。用0.3%琼脂固化。在这种植板上生长1月后,取小细胞克隆放入含一层无荷尔蒙K3培养基(K3-H)加0.2M蔗糖的塑料培养皿。用0.5%琼脂固化。培养1个月,取成活愈伤组织放在新鲜K3-H培养基加0.2M蔗糖0.5%琼脂板上。培养1月后,仍然成活的愈伤组织叫想像的转化体。把它们转入和保持在不加荷尔蒙的LS培养基,直至用生化方法测验其转化性质。
并选择取得的愈伤组织转化性质的特征是:(1)在无荷尔蒙LS上继续生长,(2)存在LpDH活性和(3)存在T-DNA。愈伤组织能在无荷尔蒙培养基生长的事实,指出它们已被转化,但有可能产生自发驯化。驯化是植物细胞在存在荷尔蒙下继代期间获得能在无荷尔蒙中生长的现象。至今用SR1原生质体和转化程序,未取得驯化组织。肿瘤专化另一标志是存在LpDH。如果愈伤组织大小达到0.5cm3,可取一小片用于检测酶活性。但不存在LpDH并不表明该组织是未转化的。转化的直接和具结论性论据,是在植物DNA中测出外源DNA或其片段,即存在T-DNA。为此可用限制性核酸内切酶解离植物材料的DNA和32P标记T-区域克隆进行Southern斑测验证实之。
(5)处理的分析
上述方法已成为用各种来源的Ti质粒DNA转化烟草原生质体的常规方法。已取得章鱼碱型Ti质粒DNA和蓝曙红型Ti质粒DNA的转化组织。但是,为了取得转化成功,不必采用整体pTi-DNA。用克隆的TL-DNA和克隆的T-DNA内部片段(HindⅢ-1)取得转化。
转化频率显然较低,即用5×105原生质体平均取得2-3转化体。通常植板在无荷尔蒙上,约3000克隆可产生必要的转化体数量。用从野生型细菌获得的章鱼碱pTi-DNA取得的转化体,观察到表型变异。可分三类:A类(植物荷尔蒙独立,LpDH正和再生负-意指再生为芽的发育);B类(植物荷尔蒙独立,LpDH负,再生负)和C类(依赖植物荷尔蒙,LpDH正,和再生负)。后一类取自K3-H培养基,已达到足以测定LpDH活性大小。在LS培养基上生长停止,除非用将愈伤组织保持在加植物荷尔蒙培养基以刺激生长。T-DNA分析证实了全部转化体核DNA中存在与Ti-质粒T-区域同质顺序。T-DNA视转化体而有差异,提出这些转化体源于独立转化事项。无论从活体或共培养取得的转化烟草组织,证实了存在着良好规定大小的T-DNA小段。由此提出T-区域可能含有特殊顺序,叫边际顺序,优先用于细菌存在时与植物DNA相整合。DNA转化中,这些顺序用处较小。事实上,用无边际顺序的克隆内部DNA片段(HindⅢ-1)试验指出,T-DNA也能整合。用上述方法产生的DNA转化体中能测得T-DNA,扩大到已知边际顺序之外,其它转化体发现较小和更混杂和分散的T-DNA。在DNA转化试验中,外源DNA的整合似比农杆菌转化T-区域给植物细胞(如共培养试验所见)的正确性较低。用完整农杆菌把T-DNA转化植物细胞试验,从未观察到已知边际顺序外的Ti质粒DNA的整合。但是,用DNA-转化体的DNA与T-区域外Ti质粒DNA的32P-标记探针杂交试验,指出即使远离T-区域的顺序一事实上从全部Ti质粒一能以整合。当用转化体DNA与32P标记小牛胸腺(用作转化试验的DNA携带者)DNA杂交时,也能测得小牛胸腺DNA与植物基因组相结合。这些结果提出DNA转化方法不限于Ti质粒DNA,一般言之,全部类型DNA都能整合,如果这种DNA上具有适合的选择标志,在转化组织中能以测得。
转化体发育产生的芽,分别继代为芽培养体。它们不生根,和无顶端优势。这是由于TDNA基因的存在而形成的特征。就此状态,这些芽与用野生型农杆菌共培养法取得的转化芽相似。经用这些芽嫁接到健旺烟草植株去顶茎上,在温室内长成成熟开花植株,它们雄性不育。用正常SR1花粉授粉后,能取得种子。指出T-DNA以孟德尔遗传方式传给子代。子代中有50%出现T-DNA。可见DNA转化后,T-DNA表现为简单显性标志。与此相反,小牛胸腺DNA几乎100%幼苗表现分离,指出这种DNA分散在转化细胞的全部染色体上。无论如何,插入的DNA顺序传给子代植株,指出它能经减数分裂而传递。这是DNA转化应用于植物遗传操作的重要条件之一。
【参考文献】:
〔1〕Hooykaas,P.J.J.and R.A.Schilperoort 1983 The molecular genetics of crown gall turnorigensis.In:Advances in Genetics(E.W.Caspari and l.G.Scandalois,eds.)pp209-283,Academic Press,New York.
〔2〕Krens,F.A.,L.Molendijk,G.J.Wullems and R.A.Schilpercoort 1982 Invitro transformation of plant protoplasts with Ti-plasmidDNA.Nature 296:72-74.
〔3〕Marton,L.G.J.Wullems,L.Molendijk and R.A.Schilpercoort 1979 In vitro transformation of cultured cells from Nicotiana tabacum by Agrobacterium tumefaciens.Nature 277:129-131.
〔4〕Memelink,J.,G.J.Wullems and R.A.Schilperooort 1983 Nopalime T-DNA is maintainedduring regeneration and generative propagation of transformed tobacco plants.Mol.Gen.genet.190:516-522.
〔5〕Wullems,G.J,L.Molendijk,G.Ooms and R.A.Schilpercoort 1981 Differential expression of crown gall tumor markers in transformants obtained after in vitro Agrobacterium tumefaciens-induced transformation of cell wall regenerating protoplasts derived from Nicotiana tabacum.Proc.Natl.Acad.Sei.U.S.A.78:4344-4348.