28.怡泼津
出处:按学科分类—医药、卫生 军事医学科学出版社《临床常用进口药物手册》第113页(91007字)
【中文释文】:
(怡泼定一号)注射用
〔产品介绍〕
红细胞生成素是一种刺激红细胞生成的糖蛋白。它是在肾脏产生,能刺激骨髓中定向红系祖细胞的分裂与分化。怡泼津是安进公司怡泼定一号的商标名,怡泼定一号被选作人红细胞生成素基因重组产品的专有名称。怡泼津是含有165氨基酸的糖蛋白,分子量为30400道尔顿,是利用DNA重组技术由引入了人红细胞生成素基因的哺乳类动物细胞产生的。它的生物作用与人体内源性的红细胞生成素相同。此产品含有与分离出的天然的红细胞生成素一样的氨基酸序列。
怡泼津制剂是一种无菌、无色、无防腐剂的液体,可由静脉注射或皮下注射。每瓶一次用的小瓶剂量有2000U,3000U,4000U或10000U的怡泼定一号。制剂用等渗的氯化钠/枸橼酸钠的缓冲液(pH值6.9±0.3)配制,内含人的血清白蛋白(2.5mg),枸橼酸钠(5.8mg),氯化钠(5.8mg),枸橼酸钠(0.06mg),以及美国药典标准的注射用水。
〔临床药理学〕
1.慢性肾衰竭病人 红细胞生成素是一种刺激红细胞生成的糖蛋白。在正常情况下,红细胞生成素在体内的生产是由组织氧合程度来调节的。一般说来,缺氧和贫血会提高红细胞生成素的产生,从而刺激红系造血。在正常人中,红细胞生成素血浆含量为0.01~0.03U/ml,而在缺氧或贫血时增至100~1000倍。相反,患有慢性肾衰竭的病人,红细胞生成素的产生受损,而这种红细胞生成素的不足就是导致贫血的主要原因。
慢性肾衰竭是指一种逐渐恶化的、通常不可逆转的肾功能衰退的临床症状。此类病人有可能出现肾功能失调的后遗症,如贫血等,但并不一定需要定期血液透析。末期的肾病病人是那些需要靠定期透析或换肾来维持生命的慢性肾衰竭病人。
已经证明,怡泼津能刺激患有慢性肾衰竭的贫血病人的红细胞生成,包括需要血液透析与不需要定期血液透析的病人。每周3次使用怡泼津后,药物效应的第一证明是10d内网状红细胞计数增加,而且通常在随后2~6周内,红细胞计数、血红蛋白和血细胞比容亦有增加。由于红细胞制造需要一段时间——从红系祖细胞到成熟再释放入循环系统需时数天——一般在两周以内观察不到具有临床意义的血细胞比容增高,有些病人可能需要长达6周才能观察到。一旦血细胞比容达到了指标范围(30%~333%),只要病人不缺铁,也没有其他疾病,怡泼津治疗便可维持这个血比容水平。
不同的病人会有不同的血细胞比容增高率,在每周3次约50~300U/kg的治疗范围内,血细胞比容增高率也与怡泼津的剂量有关。当剂量超过每周3次300U/kg时,并未见更大的生物反应。其他影响血细胞比容增高率和反应程度的因素包括铁贮存量的可用度,血细胞比容基线水平,以及并存的其他医学上因素等。
2.接受叠氮胸苷治疗的艾滋病毒感染的病人 艾滋病毒感染的病人对怡泼津的反应,取决于治疗前病人体内血清红细胞生成素的量。如果病人体内血清红细胞生成素量低于或等于500mU/ml,同时每周接受叠氮胸苷的剂量小于或等于4200mg,就可能对怡泼津治疗有反应。当病人体内血清红细胞生成素量高于500mU/ml时,对怡泼津治疗不会有反应。在连续4次共有255个病人的临床试验中,有60%~80%接受叠氮胸苷治疗的艾滋病毒感染的病人,其体内血清红细胞生成素量低于或等于500mU/ml。
这些接受叠氮胸苷治疗的艾滋病毒感染病人对怡泼津的反应是输血的需求减少,血细胞比容增高。
3.接受化学治疗的癌症病人 癌症病人的贫血可能与疾病本身有关,也可能由同时使用的化疗药物的影响所致。在接受化疗的贫血癌症病人身上已经表明,使用怡泼津治疗1个月后(即疗程的第2个月和第3个月),血细胞比容会有增加,输血的需求会减少。
对正在接受含顺铂及不含顺铂化疗的131名贫血癌症病人进行了一系列临床试验。在这些病人中,体内血清红细胞生成素的基线水平各不相同,约75%(N=83/110)的病人其体内血清红细胞生成素水平低于或等于132mU/ml,而约4%(N=4/110)的病人,其体内血清红细胞生成素水平高于500mU/ml。一般说来,血清红细胞生成素基线水平较低的病人,要比血清红细胞生成素基线水平较高的病人对怡泼津的反应更为强烈。虽然还不能定出血清红细胞生成素高过哪个具体水平,病人便不会对怡泼津有反应,但是对于血清红细胞生成素水平显着升高(例如>200mU/ml)的病人不宜推荐使用怡泼津。
4.药代动力学 静脉注射的怡泼津是以一级动力学的速率被排除,在慢性肾衰竭病人身上,循环半衰期大约是4~13h之间。在治疗剂量的范围内,血浆红细胞生成素的可测水平至少可以维持24h。给慢性肾衰竭病人皮下注射怡泼津之后,血清红细胞生成素水平是在注射后5~24h内达到高峰,然后慢慢降低。在不做透析而其血清肌酐量高于3的病人与维持做透析的病人间,半衰期并无明显差别。
正常人静脉注射怡泼津的半衰期比慢性肾衰竭病人的半衰期大约短20%左右。目前对艾滋病毒感染的病人尚未作怡泼津药物动力学的研究。
〔适应证与用法〕
1.慢性肾衰竭病人的贫血治疗 怡泼津适用于治疗由慢性肾衰竭引起的贫血症,包括进行透析的病人(末期肾病)和不进行透析的病人。怡泼津也适用于提高或维持红细胞水平(由血细胞比容或血红蛋白测量显示),并降低这些病人的输血需求。
怡泼津对严重贫血而需紧急纠正的病人并不适用。怡泼津能免除病人对维持性输血的需求,但不能取代紧急输血。
在治疗开始之前,必须先测估病人的铁贮量,包括转铁蛋白饱和度与血清铁蛋白的测定值。转铁蛋白饱和度应至少不低于20%,铁蛋白至少要有100ng/ml。在开始进行怡泼津治疗前,病人的血压必须适当控制,在治疗过程中还须严密监测和控制。有症状性贫血的非透析病人要接受治疗,其血细胞比容必须小于30%。所有接受怡泼津治疗的病人都须定期接受监测(参见“实验室监测”和“注意事项”)。
怡泼津必须在合格的医生指导下使用(参见“剂量与用法”)。
2.接受叠氮胸苷治疗的艾滋病毒感染病人的贫血治疗 怡泼津适用于接受叠氮胸苷治疗的艾滋病毒感染病人的贫血治疗。怡泼津也适用于提高或维持红细胞水平(由血细胞比容或血红蛋白测量显示),并降低这些病人的输血需求。怡泼津不适用于治疗由其他因素,如缺铁,缺叶酸,溶血或胃肠出血等导致贫血的艾滋病毒感染病人,这些病症应予另行适当处理。
当接受叠氮胸苷治疗的艾滋病毒感染的贫血病人其内源性血清红细胞生成素量小于或等于500mU/ml,而且每周接受的叠氮胸苷剂量小于或等于4200mg时,注射恬泼津100U/kg,每周3次,便能有效地降低此类病人的输血需要,并增加其红细胞量。
3.对接受化疗的癌症病人的贫血治疗 怡泼津适用于治疗非骨髓性恶性肿瘤病人因接受化疗而引起的贫血。对于将要接受化疗至少2个月的病人,恬泼津具有减少病人输血需要的作用。怡泼津不适用于治疗由其他因素,如缺铁,叶酸缺乏,溶血或胃肠出血导致贫血的癌症病人,这些病症应予另行适当的处理。
〔临床经验:对怡泼津的反应〕:
1.慢性肾衰竭病人 所有的研究表明此类病人对怡泼津的反应是一致的。在铁贮量充足的情况下(参见“治疗前铁质评估”),达到指标的血细胞比容所需的时间取决于血细胞比容基线值和血细胞比容升高。
血细胞比容的增加率取决于怡泼津的使用剂量和每个病人的不同情况。在临床试验中初始剂量50~150U/kg,每周3次,病人反应的平均血细胞比容增加率如下:
表7 血细胞比容增加
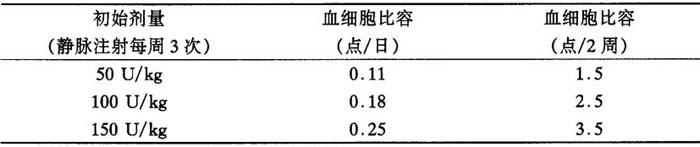
在上述剂量范围内,大约95%的病人反应出具有临床意义的血细胞比容增高,而且在大约两个月的治疗结束时,几乎所有的病人都不再需要输血。一旦血细胞比容达到指标范围,怡泼津的维持剂量便根据病人的个体情况而定。
血液透析的病人:我们进行过13次临床研究,包括总数1010个血液透析的贫血病人接受怡泼津静脉注射,疗程共达986病人年。在其中3次最大的临床试验中,维持血细胞比容在30%~36%之间所需怡泼津维持剂量的中位数值约为75U/kg(每周3次)。在美国多中心第Ⅲ期研究中,大约有65%病人需要100U/kg(每周3次)或更少的剂量,来维持其血细胞比容在35%左右。近10%的病人需要25U/kg或更少的剂量,另有10%左右的病人需要大于200U/kg(每周3次)的剂量,才能将他们的血细胞比容维持在这个水平。
不需透析的慢性肾衰竭病人:对不需透析的慢性肾衰竭病人进行了4次临床试验,涉及181个接受怡泼津治疗的病人,共约67病人年的经验。这些病人对怡泼津治疗的反应与在透析病人身上观察到的反应相似。不论怡泼津是静脉注射或皮下注射,不需透析的慢性肾衰竭病人都表现出依赖于剂量的、持久的血细胞比容增高现象,而且不论用哪种方法注射怡泼津,病人的血细胞比容的增加率是相似的。另外,怡泼津剂量在每周75~150U/kg时,便能有效地维持血细胞比容在36%~38%的水平达6个月之久。
2.接受叠氮胸苷治疗的艾滋病毒感染病人 我们对怡泼津进行过4次有安慰剂对照的临床研究,研究对象是297个同时接受叠氮胸苷治疗的贫血(血细胞比容小于30%)艾滋病毒感染病人(所有病人都接受了安进公司生产的怡泼定一号治疗)。与安慰剂对照组相比,在分组病人中(89/125用怡泼津,88/130用安慰剂),其试验前的内源性血清红细胞生成素量低于或等于50U/ml(正常的内源性血清红细胞生成素量是4~26mU/ml),怡泼津使每个病人输血的单位平均累积量减少了大约40%,对于那些需要在基线水平输血的病人,在治疗的第2个月和第3个月中,用怡泼津治疗的病人有43%变为不需要输血,而用安慰剂治疗的病人却只有18%变为不需要输血。与安慰剂对照相比,怡泼津治疗使血细胞比容显着增加。当依据第3个月中每周接受的叠氮胸苷剂量来检验治疗结果时,接受怡泼津的病人(N=51)与接受安慰剂的病人(N=54)相比,输血需要减少有统计学显着性(P<0.003)——这些病人的平均每周叠氮胸苷剂量小于或等于4200mg。在体内血清红细胞生成素量低于或等于50uU/ml,并接受i00~200U/kg怡泼津剂量(每周3次)的病人中,大约17%的人血细胞比容达到38%,他们既不需要输血,也不需要显着减少叠氮胸苷剂量。与接受安慰剂的病人的对应反应相比,在试验前体内血清红细胞生成素量高于500mU/ml的一组病人中,怡泼津治疗并不减少病人的输血需要,也不增高病人的血细胞比容。
怡泼津的疗效可能会由于治疗过程中发生的感染/炎症现象以及叠氮胸苷剂量的增加而有所减弱。因此,怡泼津的剂量必须依据这些因素来校正确定,以维持理想的红细胞生成效应。
3.接受化疗的癌症病人 我们曾在131名贫血的癌症病人中对怡泼津进行过一系列有安慰剂对照的双盲法试验。其中72名病人同时接受不含顺铂的化疗方案,59名病人同时接受含顺铂的化疗方案。病人随机分配应用怡泼津150U/kg或安慰剂,皮下注射每周3次共12周。
怡泼津治疗组病人的血细胞比容反应有明显大于相应的安慰剂处理组病人(P<0.008)(见表)。
表8 血细胞比容(%):从基线值至终值之间的平均变化*
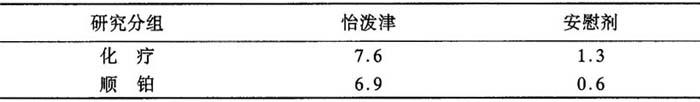
*怡泼津组病人显着高于安慰剂组病人(P<0.008)
在这两种类型的化疗研究中(怡泼津的使用剂量为150U/kg,每周3次),就治疗的第一个月后每个病人接受输血的单位数均值比较,怡泼津治疗组的病人(在第2个月和第3个月为0.71U)显着(P<0.02)低于相应的安慰剂处理组病人(在第2个月和第3个月为1.84U)。此外,就治疗的第2个月和第3个月中接受输血的病人所占比例比较(22%比43%),怡泼津治疗组比相应的安慰剂处理组显着减少(P<0.03)。
在化疗试验中,怡泼津组与安慰剂组的化疗强度是相似的,这点是由两组病人在中性粒细胞时间—曲线下有一相似面积,以及中性粒细胞绝对计数降至1.0×109/L以下的病人所占比例相似这两个现象推知。得到的证据表明,患淋巴癌和实体癌的病人对怡泼津治疗产生反应,而且不论病人有没有骨髓内肿瘤浸润,对怡泼津治疗都产生反应。
〔禁忌证〕
具有下列情形的病人应禁用怡泼津:
1.未得到控制的高血压;
2.对哺乳动物细胞衍生的产品过敏;
3.对人的血清白蛋白过敏。
〔警告〕
1.慢性肾衰竭病人
高血压:高血压未得到控制的病人不应使用怡泼津,在开始治疗前,血压应得到适当控制。在怡泼津治疗期间,通常是在治疗早期,当血细胞比容正在上升的阶段,血压可能会升高。对怡泼津有血细胞比容快速增高反应的病人(例如在任何两周期间升高4点以上),应减少怡泼津的剂量,因为血细胞比容上升过速有可能加重高血压。
癫痫发作:在接受怡泼津临床试验的慢性肾衰竭病人中,曾有癫痫发作现象发生。在血液透析的病人中,在最初90d治疗期间癫痫发作的发生率高于后来的时间(约有2.5%的病人发生)。
由于在最初90d治疗期间发生癫痫发作的潜在危险更大,因此必须密切监测血压及先兆性神经系统症状的出现。应当警告病人,在此期间要避免驾车或操作重型机器等具有潜在危险性的活动。
血栓生成:在血液透析期间,接受怡泼津治疗的病人可能需要增加肝素来加强抗凝血功能,以防止人工肾里形成凝块。血管入口内(动静脉短路)凝块的年发生率约为接受怡泼津治疗病人的每病人年0.25例。
总的说来,慢性肾衰竭病人(不论透析与否)的其他血栓性病变(例如心肌梗塞、脑血管意外、短暂性局部缺血发作等)的年发生率为怡泼津治疗的每病人年0.04例以下。对已经患有血管疾病的病人,应予以密切监测。
2.接受叠氮胸苷治疗的艾滋病毒感染病人 与慢性肾衰竭病人明显不同的是,怡泼津治疗不会引起艾滋病毒感染病人的高血压、癫痫发作和血栓性病变的加剧。
〔注意事项〕
慢性肾衰竭病人,接受叠氮胸苷治疗的艾滋病毒感染病人,以及接受化疗的癌症病人
概述:使用任何非口服生物制品时,应采取适当的警惕性措施加以护理,以防发生过敏或其他不适反应(参见“禁忌证”)。虽然在怡泼津治疗过程中偶尔观察到短暂性皮疹,但没有报告过任何严重的过敏性反应。
对于有癫痫发作病史或潜在性血液病(例如镰状细胞性贫血,骨髓增生异常综合征,或血凝过高性疾患)的病人,怡泼津治疗的安全性与有效性尚未确定。
有些女病人在接受怡泼津治疗后,月经会恢复,所以应与病人讨论怀孕的可能性,并衡量应否采取避孕措施。
血液学:在接受怡泼津治疗的慢性肾衰竭病人中偶尔观察到卟啉症加剧现象。然而,在自愿接受试验的正常人中,即使出现了迅速的红细胞生成的反应,怡泼津也并未导致尿中卟啉代谢物排泄的增加。不过,对已经患有卟啉症的病人,要谨慎使用怡泼津。
在临床前试验中,对狗和大鼠的怡泼津治疗引起了亚临床骨髓纤维化,但对猴子没有影响。骨髓纤维化是已知的人慢性肾衰竭的合并症,可能与继发性甲状旁腺机能亢进或其他未知因素有关。经过比较研究发现,一组接受怡泼津治疗12~19个月的透析病人,与一组相对应的但没有接受怡泼津治疗的病人相比,骨髓纤维化的发生率并没有增加。
慢性肾衰竭病人的血细胞比容应每周测量2次,而接受叠氮胸苷治疗的艾滋病毒感染病人和癌症病人的血细胞比容须每周测量1次,直到血细胞比容稳定下来,以后还需定期检测。
迟发或减弱的反应:如果病人对怡泼津没有反应或未能维持反应,则须考虑和评估下述病因:
1.缺铁,在铁蛋白指标正常但转铁蛋白饱和度低(少于20%)的情况下可能发生功能性缺铁,可能是因为不能快速调动贮存铁质以适应红细胞生成加速的需要。几乎所有病人最终都需要接受补充铁的治疗;
2.潜在的感染、炎症或某些恶性病情;
3.隐性失血;
4.潜在的血液病(即地中海贫血,难治性贫血,或其他骨髓增生异常症);
5.维生素不足,如叶酸或维生素B12;
6.溶血;
7.铝中毒;
8.囊状纤维性骨炎。
铁贮量测估:在怡泼津治疗前及治疗过程中,应测估病人的铁贮量,包括转铁蛋白饱和度(即血清铁除以结合铁含量)和血清铁蛋白。转铁蛋白饱和度至少应为20%,铁蛋白至少应为100ng/ml。可能需要补充铁质,来增加和维持转铁蛋白饱和度以达到足以支持怡泼津刺激的红细胞生成的水平。
药物相互作用:在临床试验中没有观察到怡泼津与其他药物发生相互作用。
致癌作用,致突变,及生育力损害:怡泼津的致癌可能性尚未被评估。怡泼津不引发细菌的基因突变(Ames试验),哺乳动物细胞的染色体畸变,小鼠体内的微核产生,或HGPRT位点的基因突变。接受100U/kg和500U/kg剂量怡泼津静脉注射的雌性大鼠,其胎儿的损耗有稍微增加的倾向。
C类怀孕:当大鼠受的剂量是人剂量的5倍时,怡泼津显示有不良反应。对孕妇尚未作过充分的对照性研究。在怀孕期间,只有证实了对胎儿的潜在好处高于潜在危险时才能使用怡泼津。
在对一组接受500U/kg剂量怡泼津的雌性大鼠进行的试验研究中,发现第一代(F1代)胎仔的体重有所下降,腹部毛发延迟出现,眼睑张开延迟,骨化延迟,以及尾椎骨数目减少。接受静脉注射怡泼津的雌性大鼠,当剂量在100U/kg和500U/kg时,其胎儿的损伤有略微增加的倾向。但是怀孕6~18d的兔子,即使接受高达500U/kg剂量的怡泼津,也无不良反应。
哺乳的母鼠:对在怀孕及哺乳期间接受500U/kg以下剂量的怡泼津的雌性大鼠的活仔(F1代)进行产后观察,并未发现有怡泼津的影响。然而一组接受500U/kg剂量怡泼津的雌性大鼠的第一代胎仔曾出现体重下降,腹部毛发及眼睑张开延迟,以及尾椎骨数目减少等现象。第二代(F2代)胎仔没有出现与怡泼津有关的影响。
怡泼津是否进人乳排出仍不得而知。由于很多药物会分泌到人乳中,故给哺乳的妇女使用怡泼津要特别谨慎。
小儿使用:小儿使用怡泼津的安全性和有效性尚未确定。
一、慢性肾衰竭病人
不需透析的慢性肾衰竭病人:对此类病人的血压和血细胞比容的监测频度不应低于需要透析维持的慢性肾衰竭病人。肾功能、液体和电解质平衡都应得到严密监测,因为有些病人可能会出现健康有所增进的感觉,以致忽略了应该开始透析的需要。
血液学:为了防止血细胞比容太快达到指标范围,或者超过指标范围(即血细胞比容30%~33%),应当遵照有关剂量及调整剂量频率的规则来使用(参见“剂量与用法”)。
使用怡泼津后血细胞比容增高很快的病人(例如在任何2周期间血细胞比容升高4点以上),需减少怡泼津剂量,因为血细胞比容上升太快有可能使高血压加重。
接受怡泼津治疗的病人的贫血被矫正后,慢性肾衰竭所特有的延长的出血时间会缩短,而趋于正常。贫血经输血矫正后,出血时间也会缩短。
在调整怡泼津剂量之前,需要有足够的时间来决定病人对某一剂量的反应。由于红细胞生成所需的时间和红细胞半衰期从每一次剂量调整(开始,增加,减少,或者停药)到血细胞比容有一个明显变化之间,可能需要间隔2~6周。
实验室检测:血细胞比容应每周测定2次,直至稳定在指标范围,并且确定了维持剂量为止。每次调整剂量之后,至少在2~6周内仍须每周2次测定血细胞比容,直到确认血细胞比容对剂量改变的反应已经稳定。此后仍应定期检测血细胞比容。
应当定期进行全血细胞计数,包括分类计数及血小板计数。在临床试验期间曾观察到血小板与白细胞计数有少量增加。虽然这些变化具有统计意义,但是并没有临床意义,数值仍保持在正常范围之内。
应当定期检测慢性肾衰竭病人的血清化学值(包括血尿素氮、尿酸、肌酐、磷和钾等)。在对透析的病人进行临床试验期间,曾观察到血尿素氮、肌酐、磷和钾有少量增加。在某些接受怡泼津治疗而不需透析的慢性肾衰竭病人中,曾观察到血清尿酸和磷有少量增加。虽然这些变化具有统计意义,但是所有数值仍属慢性肾衰竭病人的常见范围。
高血压:高血压未被控制的病人不应接受怡泼津治疗。在开始治疗前,血压应得到充分控制。所有慢性肾衰竭病人,无论是否需要透析,在接受怡泼津治疗期间,通常是在治疗早期,即血细胞比容正在上升的阶段,可能出现血压升高和高血压发作的现象。为防止高血压及其后遗症,对接受怡泼津治疗的病人要进行特殊的护理,以监视并积极控制其血压。在血细胞比容增高期间,约有25%的透析病人可能需要开始接受或加强抗高血压治疗。应该向病人讲解遵从抗高血压治疗和饮食节制的重要性。用怡泼津治疗有血细胞比容快速增高反应的病人(例如在任何两周期间升高4点以上)时,应减少怡泼津的剂量,因为血细胞比容升高过速有可能加重高血压。如果血压难以控制,怡泼津的剂量应该减少;如果临床上必要,可能要停止怡泼津治疗,直至血压重新得到控制。
癫痫发作:在接受怡泼津临床试验的慢性肾衰竭病人中,曾有癫痫发作现象发生。在透析的病人中,最初90d治疗期间发生癫痫发作的次数多于治疗后期(约有2.5%的病人发生)。
由于在最初90d治疗期间发生癫痫发作的潜在危险较大,因此必须密切监测血压及先兆性神经系统症状的出现。应当警告病人,在此期间要避免驾车或操作重型机器等具有潜在危险性的活动。
血栓生成:在血液透析期间,接受怡泼津治疗的病人可能需要增加肝素来加强抗凝血功能,以防止人工肾生成凝块。血管入口内凝块的年发生率约为怡泼津治疗的每病人年0.25例。
在接受怡泼津治疗的病人中,血细胞比容的升高与血栓形成——包括血管入口处的血栓形成的发生率之间有无关系,尚未在统计上得到确证。总的说来,慢性肾衰竭病人(不论透析与否)的其他血栓性病变(如心肌梗塞、脑血管意外、短暂性局部缺血发作等)的年发生率为怡泼津治疗的每病人年不到0.04例。对已经患有血管疾病的病人,应予以密切监测。
饮食:当血细胞比容增高,病人感觉到健康与生活状况有所好转时,应向病人再次强调遵守饮食与透析处方的重要性,特别要强调的是,高钾血症在慢性肾衰竭病人中是颇为常见的。在美国对透析病人的研究中,高钾血症的年发生率约为怡泼津治疗的每病人年发生0.11例,发病原因往往是不好好遵守药物、饮食和/或透析处方。
透析处理:怡泼津治疗所引起的血细胞比容增高和血浆容量减少,有可能影响透析的效率。在迄今为止的研究中,导致血细胞比容增高的结果似乎对透析器功能及高流量血液透析的效率都没有不良影响。在血液透析过程中,使用怡泼津的病人可能需要增加肝素以加强抗凝作用,防止人工肾生成凝结。
初期接受透析的病人可能需要多次调整透析处方。如同所有透析病人一样,接受怡泼津治疗的病人的血清化学值(包括血尿素氮,肌酐,磷和钾)应定期检测,以确定透析处方是否恰当。
肾功能:对不做透析的慢性肾衰竭病人,应密切监测其肾功能与液体及电解质平衡,因为某些病人可能会产生健康有所增进的感觉,从而掩盖了开始透析的需要。在不做透析的慢性肾衰竭病人中,有关肾功能障碍进展情况超过一年时间的研究(有安慰剂对照组)尚未完成。在对不透析的慢性肾衰竭病人较短期的试验中,接受怡泼津治疗病人的肌酐变化和肌酐清除率,与安慰剂处理的病人相比,并无显着不同。分析这些病人的血清肌酐的倒数与时间绘图的斜率,表明在怡泼津治疗开始后该斜率并无显着变化。
二、接受叠氮胸苷治疗的艾滋病毒感染病人
高血压:在对接受叠氮胸苷治疗的艾滋病毒感染病人进行怡泼津治疗时,没有观察到高血压加重的现象。但是,如果这些病人预先存在的高血压没有得到控制,则不应使用怡泼津,而且在血压得到控制之前,不应开始怡泼津治疗。在双盲法研究中,只有1位接受怡泼津治疗的病人发生过一次癫痫发作。
三、接受化疗的癌症病人
高血压:在怡泼津治疗的癌症病人中,因血细胞比容的显着增加所引起的高血压并不多见。然而,对怡泼津治疗的病人应仔细监测其血压的变化,特别是对原来就有高血压或心血管疾病历史的病人。
癫痫发作:在安慰剂对照的双盲法试验中,3.2%(N=2/63)的怡泼津治疗病人和2.9%(N=2/68)的安慰剂处理病人出现过癫痫发作。1.6%(N=1/63)的怡泼津治疗病人是在血压与血细胞比容从基线水平显着上升的过程中出现发作。不过,这2个怡泼津治疗的病人原来也都有中枢神经系统的病理变化,癫痫发作的出现可能与此有关。
血栓生成:在安慰剂对照的双盲法试验中,3.2%(N=2/63)的怡泼津治疗病人和11.8%(N=8/68)的安慰剂处理病人发生过血栓生成(如肺栓塞、脑血管意外等)。
生长因子潜能:怡泼津是一种生长因子,主要作用是刺激红细胞生成。但是,不能排除怡泼津作为生长因子,也可能刺激任何肿瘤,特别是骨髓恶性肿瘤的生长。
〔不良反应〕
一、慢性肾衰竭病人
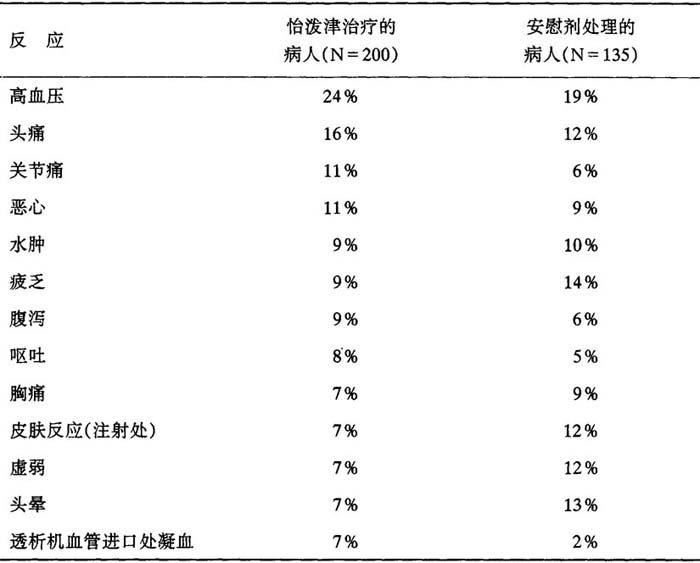
迄今为止所分析过的研究表明,病人对怡泼津一般都能很好耐受。所报告的不良反应常常为慢性肾功能衰竭的后遗症,并不一定是怡泼津治疗的后果,在安慰剂对照的双盲试验中,慢性肾功能衰竭病人总数为300人,在双盲阶段,所报告的发生率超过怡泼津治疗人数的5%,表现如下:
表9 出现反应的病人百分率
在安慰剂对照的双盲法试验中接受处理的慢性肾衰竭病人,在双盲阶段出现值得注意的重大不良反应的病人百分率如下:
癫痫发作 1.1% 1.1%
脑血管意外(CVA)/短暂性缺血性发作(TIA) 0.4% 0.6%
心肌梗塞(MI) 0.4% 1.1%
死亡 0.0% 1.7%
在美国对透析病人(超过567个病人)进行的怡泼津研究中,报告频次最高的不良反应的发生次数(每病人年发生次数)如下:高血压(0.75),头痛(0.40),心动过速(0.31),恶心/呕吐(0.26),血管入口内凝血(0.25),呼吸短促(0.14),高钾血(0.11),腹泻(0.11)。所报告的其他反应的发生率低于每病人年0.10例。
报告在使用怡泼津后几小时内即发生的反应很少,而且程度轻微,时间短暂,包括像流行性感冒那样的关节痛和肌肉痛之类症状。
在至今为止所分析过的所有研究中,无论以哪种途径使用,病人对使用怡泼津一般都能很好耐受。
过敏反应:至今还没有报告过因使用怡泼津而引起的严重的过敏反应或变态性反应。很少观察到病人有皮疹和荨麻疹的反应,而且即使有报告,也是轻微而短暂的。在迄今所测试的病人中,包括静脉注射怡泼津达2年以上的病人,都还没有迹象可以证明红细胞生成素抗体的产生。不过,如果有类似过敏症发生,应立即中断使用怡泼津,并且开始适当的治疗。
癫痫发作:怡泼津治疗与癫痫发作之间,即使有关系,这种关系也是不明确的。未接受怡泼津治疗的透析病人群体中癫痫发作发生率的基线水平难以确定,其范围看来是在每病人年5%~10%之间。在1010名接受怡泼津治疗(受药面986病人年)的透析病人中,曾发生47例癫痫发作,因此发生率约为每病人年0.048例。然而,在治疗的最初90d内,癫痫发作的发生率似乎高于随后90d期间的发生率(约有2.5%的病人发生)。虽然癫痫发作与血细胞比容升高率之间的关系还不明确,我们仍建议,如果在任何2周期间血细胞比容增高4点以上,则应减少怡泼津剂量。
高血压:多达80%的慢性肾衰竭病人有高血压病史。不论是否维持透析的慢性肾衰竭病人,在接受怡泼津治疗期间血压都可能上升。在怡泼津治疗的早期,即血细胞比容增高时期,约有25%的透析病人可能需要开始或加强抗高血压治疗。在接受怡泼津治疗的慢性肾衰竭病人中,曾观察到有高血压性脑病和癫痫发作发生。病人血压升高可能与血细胞比容的增高率有关。如果在任何两周期间血细胞比容增高4点以上,建议减少怡泼津剂量。
在临床试验中曾有血压增高的报告,通常发生在治疗的最初90d内。经分析美国多中心第Ⅲ期试验中所有病人的数据,发现一个明显的趋势:在血细胞比容增高率较快(在任何2周内增高4个血细胞比容点以上)的透析病人中,有高血压性不良反应的报告也较多。但是在1次安慰剂对照的双盲法试验中,怡泼津治疗组(150U/kg,每周3次)报告的高血压性不良反应,与安慰剂组相比并无增加。由此看来,怡泼津似乎并无直接的升压作用。应特别注意密切监测和控制接受怡泼津治疗病人的血压。
血栓生成:在血液透析期间,接受怡泼津处理的病人可能需要用肝素来增加抗凝血功能,以防止人工肾生成凝块。血管入口内凝块的年发生率约为怡泼津治疗的每病人年0.25例。
在接受怡泼津治疗的病人中,血细胞比容的升高与血栓形成——包括血管入口内(动静脉短路)的血栓反应——的发生率之间有无关系,尚未在统计上得到确证。总的说来,慢性肾衰竭病人(不论透析与否)的其他血栓性病变(如心肌梗塞、脑血管意外、短暂性局部缺血性发作等)的年发生率为怡泼津治疗的每病人年不到0.04例。对已经患有血管疾病的病人,应予以密切监测。
二、接受叠氮胸苷治疗的艾滋病毒感染病人
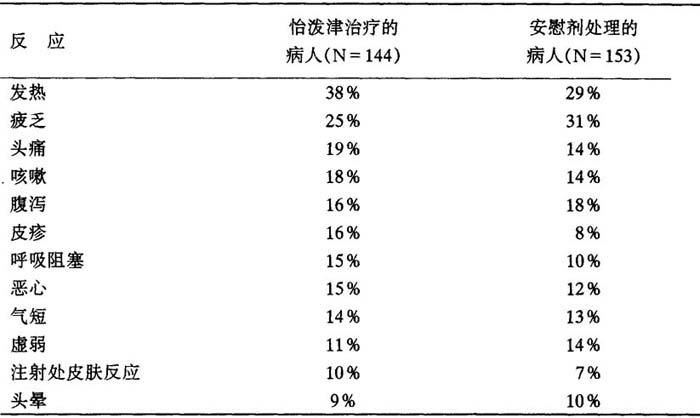
在对接受叠氮胸苷治疗的艾滋病毒感染病人进行的怡泼津临床试验中,所报告的不良反应是与病人受艾滋病毒感染的进展趋势相一致的。在安慰剂对照的双盲法研究中,对约300名接受叠氮胸苷治疗的艾滋病毒感染病人观察了3个月,发现无论在怡泼津治疗组或安慰剂处理组的病人中发生率在10%及以上的不良反应如下表:
表10 出现反应的病人百分率
两组病人之间在上述反应的发生率方面,并无统计意义上的显着差别。在297名接受研究的病人中,怡泼津没有引起机会性感染或死亡率的显着增加。在这组病人中,71名接受剂量为150U/kg每周3次的怡泼津治疗者,血清p24抗原水平似乎没有增加。物步得到的数据显示,在体外培养的感染细胞系中,怡泼津不会使人免疫缺陷病毒(HIV)的复制增加。
外周白细胞和血小板计数在怡泼津治疗后没有变化。
过敏反应:两名接受叠氮胸苷治疗的艾滋病毒感染病人,在初次接触研究药物的48h内出现了荨麻疹反应。1名病人使用怡泼津,另1名病人使用安慰剂(只含赋形剂)。2名病人对他们接受的研究药物的速发性皮肤试验都出现阳性反应,盐水对照反应则都是阴性。显然,病人对怡泼津制剂的某些成分具有预先存在的过敏反应,其原因还不清楚,但可能与艾滋病毒诱导的免疫抑制或曾经接触过某些血液制品有关。
癫痫发作:在双盲法和公开标记的怡泼津试验中,有10名接受叠氮胸苷治疗的艾滋病毒感染病人发生癫痫发作。一般说来,这些癫痫发作似乎与某些原有的病变有关,例如脑膜炎或脑瘤等,而与怡泼津治疗无关。
三、接受化疗的癌症病人
在对癌症病人使用怡泼津的临床试验中,所报告的不良反应是与病人原有的疾病状态相一致的。在安慰剂对照的双盲法研究中,对131名接受化疗的癌症病人观察了3个月,发现无论在怡泼津治疗组或安慰剂处理组的病人中发生率在10%以上的不良反应如下表:
表11 出现反应的病人百分率
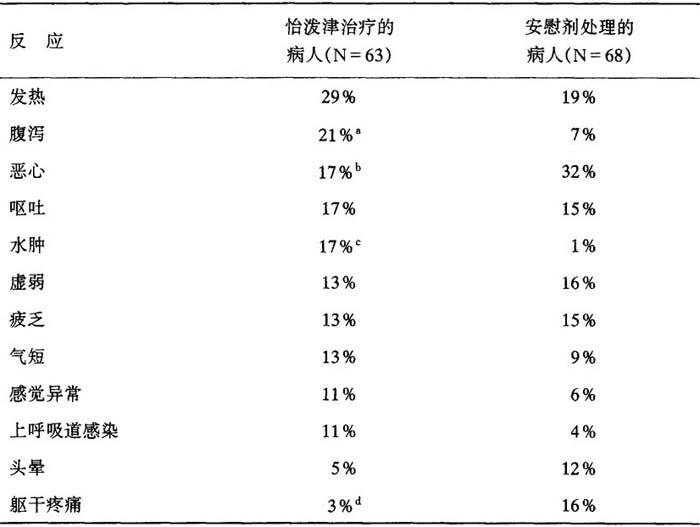
a:P=0.041 b:P=0.069 c:P=0.0016 d:P=0.017
虽然在怡泼津治疗组与安慰剂对照组的病人之间存在着一些统计意义上的差别,但怡泼津的总体安全性曲线图看来总是与晚期癌症病情进展相一致的。在怡泼津剂量高达927U/kg,疗程达32周的双盲法并随后公开标记的治疗过程中,病人(接受怡泼津全部用量的N=72)对怡泼津的不良反应的概貌,总是与晚期癌症的病情进展相一致的。
根据可以比较的存活者的数据,以及怡泼津及安慰剂处理的病人由于死亡、疾病进展或不良反应等原因而中止治疗的百分率(分别为22%和13%,P=0.25),从临床结果看来,怡泼津组和安慰剂组病人是相似的。从动物肿瘤模型和临床活体标本检查中测量实体瘤增殖对怡泼津的反应所获得的数据来看,怡泼津并不促进肿瘤生长。然而,怡泼津作为一种生长因子,可能促进某些肿瘤,特别是骨髓性肿瘤生长的可能性并不能排除。一项有对照的随机的第Ⅳ期研究正在进行之中,以进一步评估这一问题。
在怡泼律治疗后,外周白细胞计数的均值,与安慰剂对照组的相应数值比较,没有发生变化。
〔药量过度〕
一次输入或多次输入怡泼津的最大的安全剂量尚未确定。曾经用过高至1500U/kg每周3次的剂量,持续3~4周,未见任何由怡泼津本身产生的直接毒性影响。
如果不仔细监测病人的血细胞比容和适当调整剂量,用怡泼津治疗可能导致红细胞增多症。假如血细胞比容超出了指标范围,也许应暂时停用怡泼津,直至血细胞比容恢复到指标范围之后,再用较低的剂量继续怡泼津治疗(参见“剂量与用法”)。如果出现较严重的红细胞增多症,可能需采用放血来降低血细胞比容。
〔剂量与用法〕
一、慢性肾衰竭病人
经试验证明,对于慢性肾衰竭病人,怡泼津的初始用剂量50~100U/kg,每周3次,可以安全而有效地增加血细胞比容,免除对输血的依赖(参见“临床经验”)。当血细胞比容达到30%~33%的指标,或在任何2周内增高4点以上时,便应减少怡泼律的剂量。为了将血细胞比容维持在指标范围,怡泼津的剂量必须根据病人各自的情况而定。剂量的改变一般不应超过25U/kg,每周3次的范围。下表提供了一般的治疗指导方案。
初始剂量: 50~100U/kg,每周3次;静脉注射:透析病人;静脉或皮下注射:非透析慢性肾衰竭病人。
出现以下情况应减少剂量: 血细胞比容达到了指标范围;在任何2周内血细胞比容增高4点以上。
出现以下情况应增加剂量: 治疗8周后血细胞比容增加少于5~6点;而且血细胞比容低于指标范围。
维持剂量: 视各人情况具体确定。
血细胞比容指标范围: 30%~33%(最高36%)。
透析病人的怡泼津用法通常是将剂量一次静脉注射,每周3次。虽然怡泼津的用法与透析程序并无关系,但是可以在透析程序结束时将怡泼津注入静脉管,这样可避免再次插静脉管。对不透析的慢性肾衰竭病人,怡泼津可以静脉注射,也可以皮下注射。
治疗期间应定期检测各种血液参数(参见“实验室检测”)。
治疗前铁质测估:在怡泼津治疗之前与治疗期间,应测估病人的铁贮量,包括转铁蛋白饱和度(血清铁除以铁结合容量)和血清铁蛋白的测定。转铁蛋白饱和度至少应为20%,铁蛋白至少为100ng/ml。病人可能需要补充铁质,来增加和维持转铁蛋白饱和度达到足以支持怡泼津刺激的红细胞生成所需的水平。
1.剂量调整 当血细胞比容达到30%~33%时,怡泼津的剂量应减少约25U/kg,每周3次,以防止超过指标范围。一旦血细胞比容达到指标范围,必须按病人各自的情况来确定维持剂量(参见“维持剂量”)。
在任何时候,如果血细胞比容在2周内增高了4点以上,应立即减少怡泼津的剂量。在减少剂量之后,必须在2~6周内每周2次检测血细胞比容,并依照“维持剂量”一节介绍的方法进一步调整剂量。
当血细胞比容接近或者超过36%时,应暂时停用怡泼津,直到血细胞比容降低到30%~33%的指标范围。在重新开始治疗时,怡泼津的剂量应减少约25U/kg,每周3次。
如果在治疗8周之后血细胞比容的增高不到5~6点,而且铁贮量还充足(参见“迟发或减弱的反应”),怡泼津的剂量可按25U/kg,递增每周3次量。以后可每隔4~6周再增加25U/kg,每周3次,直至获得理想的反应。
2.维持剂量 维持剂量必须视病人各自的情况而定。当血细胞比容接近或者超过36%时,应暂时停用怡泼津,直到血细胞比容降低到33%或以下。在重新开始治疗时,怡泼津的剂量应减少约25U/kg,每周3次,或略去给药次数,并应给予适当的时间间隔(即2~6周)使反应稳定下来。
如果血细胞比容停留于或降低到指标范围以下,应重新测估铁贮量。如果转铁蛋白饱和度低于20%,则应当补充铁质。如果转铁蛋白饱和度高于20%,可能应将怡泼津的剂量增加25U/kg,每周3次。这样的剂量增加频率不应超过每月1次,除非在临床上指示有增加的必要,因为血细胞比容对剂量增加的反应时间可能需要2~6周。在剂量增加后2~6周内,应每周2次检测血细胞比容。
在美国多中心第Ⅲ期试验中,血液透析病人的维持剂量中值是75U/kg每周3次,其中约65%的病人需要100U/kg每周3次或较少的剂量,来维持其血细胞比容在32%~38%范围内(维持剂量的范围是从12.5U/kg到525U/kg,每周3次)。近10%的病人需要25U/kg或更少的剂量,另外约10%的病人需要200U/kg(每周3次)以上的剂量,来维持其血细胞比容在这个范围内。
不透析的慢性肾衰竭病人的维持剂量也须视病人各自的情况而定。经试验证明,每周75~150U/kg的怡泼津剂量可维持36%~38%的血细胞比容达6个月。
3.迟发或减弱的反应 在开始怡泼津治疗约2个月内,95%以上的慢性肾衰竭病人出现了具有临床意义的血细胞比容增高的反应,而且几乎所有病人都不再需要依赖输血。
如果病人没有出现反应,或未能维持反应,则应考虑其他病因,并根据临床征象作出评估。参见“注意事项”一节有关迟发或减弱反应的讨论。
二、接受叠氮胸苷治疗的艾滋病毒感染病人
在开始怡泼津治疗之前,建议先测定体内血清红细胞生成素水平(输血前)。从已有的证据可以看出,接受叠氮胸苷治疗的艾滋病毒感染病人,若其体内血清红细胞生成素水平高于500mU/ml,便不大可能对怡泼津治疗产生反应。
1.初始剂量 对于血清红细胞生成素量等于或低于500mU/ml,所接受的叠氮胸苷剂量等于或少于每周4200mg的病人,推荐的怡泼津初始剂量为100U/kg,每周3次,静脉或皮下注射,持续8周。
2.增加剂量 在治疗的剂量调整阶段,应每周监测血细胞比容。如果在治疗8周后,从减少输血需求或增加血细胞比容方面考虑,病人的反应并不令人满意,可将怡泼津的剂量增加50~100U/kg,每周3次。此后应每隔4~8周评价病人的反应,并按照50~100U/kg每周3次的递增量调整剂量。如果病人对300U/kg每周3次的怡泼津剂量仍无令人满意的反应,那么很可能他们对更高剂量的怡泼津也不会有反应了。
3.维持剂量 在达到理想的反应(即减少输血需求或增加血细胞比容)之后,应该根据叠氮胸苷剂量的变化及出现间发性感染或炎症等因素来校正制定怡泼津的剂量,以维持这种反应。如果血细胞比容超过40%,应当中断该剂量,直至血细胞比容下降到36%。当治疗恢复时,剂量应减少25%,然后精确调整到可以维持理想的血细胞比容值。
三、接受化学治疗的癌症病人
在接受这些试验的病人中,体内血清红细胞生成素的基线水平各不相同,约75%(N=83/110)病人的体内血清红细胞生成素水平低于132mU/ml,约4%(N=4/110)的病人高于500mU/ml。一般说来,血清红细胞生成素水平较低的病人,要比基线水平较高的病人对怡泼津的反应更为强烈。虽然还不能确定血清红细胞生成素高过哪一水平,病人便可能不会对怡泼津治疗有反应,但是对于血清细胞生成素水平显着升高(例如超过200mU/ml)的病人,则不推荐使用怡泼津。对接受怡泼津治疗的病人应每周检测其血细胞比容,直至血细胞比容达到稳定。
1.初始剂量 推荐的怡泼津初始剂量为150U/kg,每周3次,皮下注射。
2.剂量调整 如果在治疗8周后,从减少输血需求或增加血细胞比容方面考虑,病人的反应并不令人满意时可将怡泼津的剂量增加到300U/kg,每周3次。如果病人对300U/kg每周3次的怡泼津剂量仍无令人满意的反应,那么很可能他们对更高剂量的怡泼津也不会有反应了。如果血细胞比容超过40%,怡泼津剂量应当停止,直至血细胞比容下降到36%。当治疗重新恢复时,怡泼津的剂量应减少25%,并精确调整到可以维持理想的血细胞比容值。如果怡泼津的初始剂量即能引起非常迅速的血细胞比容反应(如在任何2周内增高了4个百分点以上)就应该减少怡泼津的剂量。
〔怡泼津制剂与用法〕
1.切勿振摇。振摇可能会使糖蛋白质变性,导致其失去生物活性。
2.凡属非口服的药物产品,在使用前都应用肉眼检验有无颗粒物质及变色。不可使用任何呈现颗粒物质或变色的药瓶。
3.采用无菌技术,用消毒针连接消毒注射器。拉掉怡泼津药瓶的拉盖,用消毒剂擦拭瓶膜。将针头插入药瓶,然后将适量药液抽进注射器内。
4.每瓶只用1剂,不要再将针头插入药瓶。剩余部分要扔掉。本药品不含防腐剂。
5.不要与其他药液混合注射。
〔供应方式〕
怡泼津是以小瓶瓶装供应,每瓶1.0ml,内含2000(NDC55513-126-01)、3000(NDC55513-267-01)、4000(NDC5551-148-01)或10000(NDC55513-144-01)单位的无菌、无防腐剂的怡泼津一号溶液。每种剂量以盒供应,每盒装有10支一次使用的小瓶。
〔贮存〕
贮存温度为2~8℃。切勿冰冻或振摇。
〔生产厂家〕
美国加利福尼亚州千橡市安进公司
【外文释文】:
For Injection
Descripition
Erythropoietin is a glycoprotein which stimulates red blood cell production.It is produced in the kidney and stimulates the division and differentiation of committed erythroid progenitors in the bone marrow.Epogen is the Amgen Inc.trademark for Epoetin alfa which has been selected as the proper name for recombinant human erythropoietin.
Epogen,a 165 amino acid glycoprotein manufactured by recombinant DNA technology,has the same biological effects as endogenous erythropoietin.It has a molecular weight of 30,400 daltons and is produced by mammalian cells into which the human erythropoietin gene has been introduced.The product contains the identical amino acid sequence of isolated natural erythropoietin.
Epogen is formulated as a sterile,colorless,preservative-free liquid for intravenous or subcutaneous administration.Each single-use vial contains 2 000,3 000,4 000,or 10,000 units of Epoetin alfa formulated in an isotonic sodium chloride/sodium citrate buffered solution (pH 6.9±0.3)containing Albumin(Human)(2.5mg),sodium citrate(5.8mg),sodium chloride(5.8mg),and citric acid(0.06mg)in Water for Injection,USP.
Clinical pharmacology
Chronic renal failure patients:Erythropoietin is a glycoprotein which stimulates red blood cell production.Endogenous production of erythropoietin is normally regulated by the level of tissue oxygenation.Hypoxia and anemia generally increase the production of erythropoietin,which in turn stimulates erythropoiesis.In normal subjects,plasma erythropoietin levels range from 0.01 to 0.03 U/ml,and increase up to 100-to 1 000-fold during hypoxia or anemia.In contrast,in patients with chronic renal failure(CRF),production of erythropoietin is impaired,and this erythropoietin deficiency is the primary cause of their anemia.
Chronic renal failurs is the clinical situation in which there is a progressive and usually irreversible decline in kidney function.Such patients may manifest the sequelae of renal dysfunction,including anemia,but do not necessarily require regular dialysis.Patients with end-stage renal disease(ESRD)are those patients with CRF who require regular dialysis or kidney transplantation for survival.
Epogen has been shown to stimulate erythropoiesis in anemic patents with CRF,including both patients on dialysis and those who do not require regular dialysis.The first evidence of a response to the three times weekly(T.I.W.)administration of epogen is an increase in the reticulocyte count within 10 days,followed by increases in the red cell count,hemoglobin,and hematocrit,usually within 2-6 weeks.(Because of the length of time required for erythropoiesis-several days for erythroid progenitors to mature and be released into the circulation-a clinically significant increase in hematocrit is usually not observed in less than 2 weeks and may require up to 6 weeks in some patients).Once the hematocrit reaches the target range(30%-33%),that level can be sustained by epogen therapy in the absence of iron deficiency and concurrent illnesses.
The rate of hematocrit increase varies between patients and is dependent upon the dose of epogen,within a therapeutic range of approximately 50~300 U/kg T.I.W.a greater biologic response is not observed at doses exceeding 300 U/kg T.I.W.Other factors affecting the rate and extent of response include availability of iron stores,the baseline hematocrit,and the presence of concurrent medical problems.
Zidovudine-treated HIV-infected patients:
Responsiveness to epogen in HIV-infected patients is dependent upon the endogenous serum erythropoietin level prior to treatment.Patients with endogenous serum erythropoietin levels≤500 U/l,and who are receiving a dose of zidovudine≤4 200 mg/week,may respond to epogen therapy.
Patients with endogenous serum erythropoietin levels>500 U/l do not appear to respond to epogen therapy.In a series of four clinical trials involving 255 patients,60-80% of HIV-infected patients treated with zidovudine had endogenous serum erythropoietin levels ≤500 U/l.
Response to epogen in zidovudine-treated HIV-infected patients is manifested by reduced transfusion requirements and increase hematocrit.
Cancer patients on chemotherapy:
Anemia in cancer patients may be related to the disease itself or the effect of concomitantly administered chemotherapeutic agents.
Epogen has been shown to increase hematocrit and decrease transfusion requirements after the first month of therapy,(Months 2 and 3 of therapy),in anemic cancer patients undergoing chemotherapy.
A series of clinical trials enrolled 13 1 anemic cancer patients who were receiving cyclic cisplatin or non cisplatin-containing chemotherapy.Endogenous baseline serum erythropoietin levels varied among patients in these trials with approximately 75%(N=83/1 10)having endogenous serum erythropoietin levels≤132 U/l,and approximatel 4%(N=4/110)of patients having endogenous serum erythropoietin levels>500 U/l.In general,patients with lower baseline serum erythropoietin levels responded more vigorously to epogen than patients with higher baseline serum erythropoietin levels.Although no specific serum erythropoietin level can be stipulated above which patients would be unlikely to respond to Epogen therapy,treatment of patients with grossly elevated serum erythropoietin levels(e.g.,>200 U/l)is not recommended.
Pharmacokinetics:
Intravenously administered epogen is eliminated at a rate consistent with first order kinetics with a circulating half-life ranging from approximately 4-13 hours in patients with CRF.Within the therapeutic dose range,detectable levels of plasma erythropoietin are maintained for at least 24 hours.After subcutaneous administration of epogen to patients with CRF,peak serum levels are achieved within 5-24 hours after administration and decline slowly thereafter.There is no apparent difference in half-life between patients not on dialysis whose serum creatinine levels were greater than 3,and patients maintained on dialysis.
In normal volunteers,the half-life of intravenously administered epogen is approximately 20%shorter than the half-life in CRF patients.The pharmacokinetics of epogen have not been studied in HIV-infected patients.
Indications and Usage
Treatment of anemia of chronic renal failure patients:
Epogen is indicated in the treatment of anemia associated with chronic renal failure,including patients on dialysis(end-stage renal disease)and patients not on dialysis.Epogen is indicated to elevate or maintain the red blood celllevel(as manifested by the hematocrit or hemoglobin determinations)and to decrease the need for transfusions in these patients.
Epogen is not intended for patients who require immediate correction of severe anemia.Epogen may obviate the need for maintenance transfusions but is not a substitute for emergency transfusion.
Prior to initiation of therapy,the patient’s iron stores,including transferrin saturation and serum ferritin,should be evaluated.Transferrin saturation should be at least 20%and ferritin at least 100 ng/ml.Blood pressure should be adequately controlled prior to initiation of epogen therapy,and must be closely monitored and controlled during therapy.Non-dialysis patients with symptomatic anemia considered for therapy should have a hematocrit less than 30%.All patients on epogen therapy should be regularly monitored(see“Laboratory Monitoring”and“Precautions”).
Epogen should be administered under the guidance of a qualified physician(see“Dosage and Administration”).
Treatment of anemia in zidovudime-treated HIV..infectec patients:
Cated for the treatment of anemia related to therapy with zidovuine in HIV-Infected pa
tients.Epogen is indicated to elevate or maintain the red blood cell level as manifested by the hematocrit or hemoglobin determinations)and to decrease the need for transfusions in these patients.Epogen is not indicated for the treatment of anemia in HIV-infected patients due to other factors such as iron or folate deficeies,hemolysis or gastrointestinal bleeding which should be managed appriately.
Epogen at a dose of 100 U/kg three times per week,is effective in decreasing the transfusion reguirement and increasing the red blood cell level of anemic,HIV-infected Patients treated with zidovudine,when the endogenous serum erythropoietin levelis≤500 U/ml and when patients are receiving a dose of zidovudine≤4 200mg/week.
Treatment of anemia in cancer patients on chemotherapy:
Epogen is indicated for the treatment of anemia in patients with non-myeloid malignancies where anemia is due to the effect of concomitantly administered chemotherapy.Epogen is indicated to decrease the need for transfusions in patients who will be receiving concomitantchemo-therapy for a minimum of 2 months.Epogen is not indicated for the treatment of anemia in cancer patients due to other factors such as iron or folate deficiencies,hemolysis or gastrointestinal bleeding which should be managed appropriately.
Clinical Experience:Response to Epogen:
Chronic renal failure patients:
Response to epogen was consistent across all studies.In the presence of adequate iron stores(see“Pretherapy iron evaluation”),the time to reach the target hematocrit is a function of the baseline hematocrit and the rate of hematocrit rise.
The rate of increase in hematocrit is dependent upon the dose of epogen administered and individual patient variation.In clinical trials at starting doses of 50-150 U/kg T.I.W.,patients responded with an average rate of hematocrit rise of:
Table 7 hematocrit increase

Over this dose range,approximately 95%of all patients responded with a clinically significant increase in hematocrit,and by the end of approximately 2 months of therapy virtually all patients were transfusion-independent.Once the target hematocrit was achieved,the maintenance dose was individualized for each patient.
Patients on Dialysis:Thirteen clinical studies were conducted,involving intravenous administration to a total of 1 010 anemic patients on dialysis for 986 patient-years of epogen therapy.In the three largest of these clinical trials,the median maintenance dose necessary to maintain the hematocrit between 30%-36% was approximately 75 U/kg(T.I.W.).In the U.S.multicenter Phase Ⅲ study,approximately 65% of the patients required doses of 100 U/kg T.I.W.,or less,to maintain their hematocrit at approximately 35%.Almost 10% of patients required a dose of 25 U/kg,or less,and approximately 10% required a dose of more than 200 U/kg T.I.W.to maintain their hematocrit at this level.
Patients with CRF Not Requiring Dialysis:Four clinical trials were conducted in patients with CRF not on dialysis involving 181 epogen-treated patients for approximately 67 patientyears of experience.These patients responded to epogen therapy in a manner similar to that observed in patients on dialysis.Patients with CRF not on dialysis demonstrated a dose-dependent and sustained increase in hematocrit when epogen was administered by either an intravenous(IV)or subcutaneous(SC)route,with similar rates of rise of hematocrit when epogen was administered by either route.Moreover,epogen doses of 75-150 U/kg per week have been shown to maintain hematocrits of 36%-38% for up to 6 months.
Zidovudine-treated HIV-infected patients:Epogen has been studied in four placebo-controlled trials enrolling 297 anemic(hematocrit<30%)HIV-infected(AIDS)patients receiving concomitant therapy with zidovudine(all patients were treated with Epoetin alfa manufactured by Amgen Inc.).In the subgroup of patients(89/125 epogen,and 88/130 placebo)with prestudy endogenous serum erythropoietin levels≤500 U/l(normal endogenous serum erythropoietin levels are 4-26 U/I),epogen reduced the mean cumulative number of units of blood transfused per patient by approximately 40%,as compared to the placebogroup.Among those patients who required transfusions at baseline,43% of epogen-treated patients versus 18% of placebo-treated patients were transfusion-independent during the seeond and third months of therapy.Epogen therapy also resulted in significant increases in hematocrit in comparison to placebo.When examining the results according to the weeklydose of zidovudine received during Month 3 of therapy,there was a statistically significant(P<0.003)reduction in transfusion requirements in epogen-treated patients(N=5 1)compared to placebo treatd patients(N=54)whose mean weekly zidovudine dose was≤4 200mg/week.Approximately 17% of the patients with endogenous serum erythropoietin levels ≤500 U/L receiving epogen in doses from 100-200 U/kg three times weekly(T.I.W.)achieved a hematocrit of 38% without administration of transfusions or significant reduction in zidovudine dose.In the subgroup of patients whose prestudy endogenous serum erythropoietin levels were>500 U/l,epogen therapy did not reduce transfusion requirements or increase hematocrit,compared to the corresponding responses in placebo-treated patients.
Responsiveness to epogen therapy may be blunted by intercurrent infectious/inflam-matory episodes and by an increase in zidovudine dosage.Consequently,the dose of epogen must be titrated based on these factors to maintain the desired erythropoietic response.
Cancer patients on chemotherapy:Epogen has been stuided in a series of placebo··controlled,double-blind trials in a total of 131 anemic cancer patients.Within this group,72 patients were treated with concomitant non-cisplatin-containing chemotherapy regimens.and 59 patients were treated with concomitant cisplatin-containing chemotherapy regimens.Patients were randomized to epogen 150 U/kg or placebo subcutaneously T.I.W.for 12 weeks.
Epogen therapy was associated with a significantly(P<0.008)greater hematocrit response than in the corresponding placebo-treated patients(see Table).
Talbe 8 Hematocrit(%):mean change from baseline to final value*

*Significantly higher in epogen patients than in placebo patients(P<0.008)
In the two types of chemotherapy studies,(utilizing an epogen dose of 150 U/kg T.I.W.),the mean number of units of blood transfused per patient after the first month of therapy was significantly(P<0.02)lower in epogen-treated patients(0.71 U in Months 2,3)than in corresponding placebo-treated patients(1.84 U in Months 2,3).Moreover,the proportion of patients transfused during Months 2 and 3 of therapy combined was significantly(P<0.03)lower in the epogen-treated patients than in the corresponding placebo-treated patients(22% versus 43%).
Comparable intensity of chemotherapy in the epogen and placebo groups in the chemotherapy trials was suggested by a similar area under the neutrophil time curve in epogen-and placebo-treated patients as well as by a similar proportion of patients in epogen··and placebo-treated groups whose absolute neutrophil counts fell below 1 × 109cells/I.Availableevidence suggests that patients with lymphoid and solid cancers respond to epogen therapy,and that patients with or without tumor infiltration of the bone marrow respond to epogen therapy.
Contraindications
Epogen is contraindicated in patients with:
1.Uncontrolled hypertension.
2.Known hypersensitivity to mammalian cell-derived products.
3.Known hypersensitivity to Albumin(Human).
Warnings
Chronic renal failure patients
Hypertension:Patients with uncontrolled hypertension should not be treated with epogen;blood pressure should be controlled adequately before initiation of therapy.Blood pressure may rise during epogen therapy,often during the early phase of treatment when the hematocrit is increasing.
For patients who respond to epogen with a rapid increase in hematocrit(e.g.,more than 4 points in any 2-week period),the dose of epogen should be reduced because of the possible association of excessive rate of rise of hematocrit with an exacerbation of hypertension.
Seizures:Seizures have occurred in patients with CRF participating in epogen clinical trials.
In patients on dialysis,there was a higher incidence of seizures during the first 90 days of therapy(occurring in approximately 2.5% of patients),as compared with later timepoints.
Given the potential for an increased risk of seizures during the first 90 days of therapy,blood pressure and the presence of premonitory neurologic symptoms should be monitored closely.Patients should be cautioned to avoid potentially hazardous activities such as driving or operating heavy machinery during this period.
Thrombotic Events:During hemodialysis,patients treated with epogen may require increased anticoagulation with heparin to prevent clotting of the artificial kidney.Clotting of the vascular access(A-V shunt)has occurred at an annualized rate of about 0.25 events per patient-year on epogen therapy.
Overall,for patients with CRF(whether on dialysis or not),other thrombotic events(e.g.,myocardial infarction,cerebrovascular accident,transient ischemic attack)have occurred at an annualized rate of less than 0.04 events per patient-year of epogen therapy.
Patients with pre-existing vascular disease should be monitored closely.
Zidovudine-Treated HIV-Infected Patients:In contrast to CRF patients,epogen therapy has not been linked to acceleration of hypertension,seizures,and thrombotic events in HIV-infected patients.
Precautions
Chronic renal failure patients,Zidovudine-treated HIV-infected patients,and cancer patients on chemotherapy
General:The parenteral administration of any biologic product should be attended by appropriate precautions in case allergic or other untoward reactions occur(see“Contraindications”).While transient rashes have occasionally been observed concurrently with epogen therapy,no serious allergic or anaphylactic reactions have been reported.
The safety and efficacy of epogen therapy have not been established in patients with a known history of a seizure disorder or underlying hematologic disease(e.g.,sickle cell anemia,myelodysplastic syndromes,or hypercoagulable disorders).
In some female paients,menses have resumed following epogen therapy;the possibility of potential pregnancy should be discussed and the need for contraception evaluated.
Hematology:Exacerbation of porphyria has been observed rarely in epogen-treated patients with CRF.However,epogen has not caused increased urinary excretion of porphyrinmetabolites in normal volunteers,even in the presence of a rapid erythropoietic response.Nevertheless,epogen should be used with caution in patients with known porphyria.
In pre-clinical studies in dogs and rats,but not in monkeys,epogen therapy was associated with subclinical bone marrow fibrosis.Bone marrow fibrosis is a known complication of CRF in humans and may be related to secondary hyperparathyroidism or unknown factors.The incidence of bone marrow fibrosis was not increased in a study of patients on dialysis who were treated with epogen for 12-19 months,compared to the incidence of bone marrow fibrosis in a matched group of patients who had not been treated with epogen.
Hematocrit in CRF patients should be measured twice a week;zidovudine-treated HIVinfected,and cancer patients should have hematocrit measured once a week until hematocrit has been stabilized,and measured periodically thereafter.
Delayed or diminished response:If the patient fails to respond or to maintain a response,the following etiologies should be considered and evaluated:
1.Iron deficiency:functional iron deficiency may develop with normal ferritin levels,but low transferrin saturation(less than 20%),presumably due to the inability to mobilize iron stores rapidly enough to support increased erythropoiesis.Virtually all patients will eventually require supplemental iron therapy.
2.Underlying infectious,inflammatory,or malignant processes.
3.Occult blood loss.
4.Underlying hematologic diseases(i.e.,thalassemia,refractory anemia,or other myelodysplastic disorders).
5.Vitamin deficiencies:folic acid or vitamin B12.
6.Hemolysis.
7.Aluminum intoxication.
8.Osteitis fibrosa cystica.
Iron evaluation:Prior to and during epogen therapy,the patient’s iron stores,including transferrin saturation(serum iron divided by iron binding capacity)and serum ferritin,should be evaluated.Transferrin saturation should be at least 20%,and ferritin should be at least 100 ng/ml.Supplemental iron may be required to increase and maintain transferrin saturation to levels that will adequatly support epogen-stimulated erythropoiesis.
Drug Interaction:No evidence of interaction of epogen with other drugs was observed in the course of clinical trials.
Carcinogenesis,Mutagenesis,and Impairment of Fertility:Carcinogenic potential of epogen has not been evaluated.Epogen does not induce bacterial gene mutation(Ames Test),chromosomal aberrations in mammalian cells,micronuclei in mice,or gene mutation at the HGPRT locus.In female rats treated intravenously with epogen,there was a trend for slightly increased fetal wastage at doses of 100 U/kg and 500 U/kg.
Pregnancy category C:epogen has been shown to have adverse effects in rats when given in doses five times the human dose.There are no adequate and well-controlled studies in pregnant women.Epogen should be used during pregnancy only if potential benefit justifies the potential risk to the fetus.
In studies in female rats,there were decreases in body weight gain,delays in appearance of abdominal hair,delayed eyelid opening,delayed ossification,and decreases in the number of caudal vertebrae in the F1 fetuses of the 500 U/kg group.In female rats treated intravenously,there was a trend for slightly increased fetal wastage at doses of 100 and 500 U/kg.Epogen has not shown any adverse effect at doses as high as 500 U/kg in pregnant rabbits(from day 6-18 of gestation).
Nursing Mothers:Postnatal observations of the live offspring(F1 generation)of female rats treated with epogen during gestation and lactation revealed no effect of epogen at doses of up to 500 U/kg.There were,however,decreases in body weight gain,delays in appearance of abdominal hair,eyelid opening,and decreases in the number of caudal vertebrae in the F1 fetuses of the 500 U/kg group.There were no epogen-related effects on the F2 generation fetuses.
It is not known whether epogen is excreted in human milk.Because many drugs are excreted in human milk,caution should be exercised when epogen is administered to a nursing Woman.
Pediatric use:The safety and effectiveness of epogen in children have not been established.
Chronic Renal Failure Patients
Patients with CRF not requiring dialysis:Blood pressure and hematocrit should be monitored no less frequently than for patients maintained on dialysis.Renal funcuion and fluid and electrolyte balance should be closely monitored,as an improved sense of well-being may obscure the need to initiate dialysis in some patients.
Hematology:In order to avoid reaching the target hematocrit too rapidly.or exceeding the target range(hematocrit of 30%-33%),the guidelines for dose and frequency of dose adjustments(see“Dosage and Administration”)should be followed.
For patients who respond to epogen with a rapid increase in hematocrit(e.g.,more than 4 points in any 2-week period),the dose of epogen should be reduced because of the possible association of excessive rate of rise of hematocrit with an exacerbation of hypertension.
The elevated bleeding time characteristic of CRF decreases toward normal after correction of anemia in epogen-treated patients.Reduction of bleeding time also occurs after correction of anemia by transfusion.
Sufficient time should be allowed to determine a patient’s responsiveness to a dosage of epogen before adjusting the dose.Because of the time required for erythropoiesis and the red cell half-life,and interval of 2-6 weeks may occur between the time of a dose adjustment(initiation,increase,decrease,or discontinuation)and a significant change in hematocrit.
Laboratory monitoring:The hematocrit should be determined twice a week until it has stabilized in the target range and the maintenance dose has been established.After any dose adjustment,the hematocrit should also be determined twice weekly for at least 2-6 weeks until it has been determined that the hematocrit has stabilized in response to the dose change.The hematocrit should then be monitored at regular intervals.
A complete blood count with differential and platelet count should be performed regularly.During clinical trials,modest increases were seen in platelets and white blood cell counts.While these changes were statistically significant,they were not clinically significant and the values remained within normal ranges.
In patients with CRF,serum chemistry values[including blood urea nitrogen(BUN),uric acid,creatinine,phosphorus,and potassium]should be monitored regularly.During clinical trials in patients on dialysis,modest increases were seen in BUN,creatinine,phosphorus,and potassium.In some patients with CRF not on dialysis,treated with epogen,modest increases in serum uric acid and phosphorus were observed.While changes were statistically significant,the values remained within the ranges normally seen in patients with CRF.
Hypertension:Patients with uncontrolled hypertension should not be treated with epogen;blood pressure should be controlled adequately before initiation of therapy.Blood pressure may rise and episodes of hypertension may increase during epogen therapy in all CRF patients,whether or not they require dialysis,often during the early phase of treatment when the hematocrit is increasing.To prevent hypertension and sequelae,particular care needs tobe taken in patients treated with epogen to monitor and aggressively control blood pressure.During the period when hematocrit is increasing,approximately 25% of patients on dialysis may requiire iiniitiiatiion of,or increases in,antihypertensive therapy.Patients should be advised as to the importance of compliance with antihypertensive therapy and dietary restrictions.For patients who respond to epogen with a rapid increase in hematocrit(e.g.,more than 4 points in any 2-week period),the dose of epogen should be reduced because of the possible association of excessive rate of rise of hematocrit with an exacerbation of hypertension.If blood pressure is difficult to control,the dose of epogen should be reduced;if clinically indicated,epogen may be withheld until blood pressure control is re-established.
Seizures:Seizures have occurred in patients with CRF participating in epogen clinical trials.In patients on dialysis,there was a higher incidence of seizures during the first 90 days of therapy(occurring in approximately 2.5% of patients),as compared with later timepoints.
Given the potential for an increased risk of seizures during the first 90 days of therapy,blood pressure and the presence of premonitory neurologic symptoms should be monitored closely.Patients should be cautioned to avoid potentially hazardous activites such as driving or operating heavy machinery during this period.
Thrombotic events:During hemodialysis,patients treated with epogen may require increased anticoagulation with heparin to prevent clotting of the artificial kindney.Clotting of the vascular access has occurred at an annualized rate of about 0.25 events per patient-year on epogen therapy.
A relationship has not been established with statistical certainty between a rise in hematocrit and the rate of thrombotic events[including thrombosis of vascular access(A-V shunt)]in epogen-treated patients.Overall,for patients with CRF(whether on dialysis or not),other thrombotic events(e.g.,myocardial infarction,cerebrovascular accident,transient ischemic attack)have occurred at an annualized rate of less than 0.04 events per patient-year of epogen therapy.Patients with pre-existing vascular disease should be monitored closely.
Diet:As the hematocrit increases and patients experience an improved sense of wellbeing and quality of life,the importance of compliance with dietary and dialysis prescrip tions should be reinforced.In particular,hyperkalemia is not uncommon in patients with CRF.In U.S.studies in patients on dialysis,hyperkalemia has occurred at an annualized rate of approximately 0.1 1 episodes per patient-year of epogen therapy,often in association with poor compliance to medication,dietary and/or dialysis prescriptions.
Dialysis management:Therapy with epogen results in an increase in hematocrit and adecrease in plasma volume which could affect dialysis efficiency.In studies to date,the resulting increase in hematocrit did not appear to adversely affect dialyzer function or the efficiency of high flux hemodialysis.During hemodialysis,patients treated with epogen may require increased anticoagulation with heparin to prevent clotting of the artificial kidney.
Patients who are marginally dialyzed may require adjustments in their dialysis prescription.As with all patients on dialysis,the serum chemistry values[including blood urea nitogen(BUN),creatinine,phosphorus,and potassium]in epogen-treated patients should be monitored regularly to assure the adequacy of the dialysis prescription.
Renal function:In patients with CRF not on dialysis,renal function and fluid and electrolyte balance should be closely monitored,as an improved sense of well-being may obscure the need to initiate dialysis in some patients.In patients with CRF not on dialysis,placebocontrolled studies of progression of renal dysfunction over periods of greater than one year have not been completed.In shorter-term trials in patients with CRF not on dialysis,changes in creatinine and creatinine clearance were not significantly different in epogen..treated patients,compared with placebo-treated patients.Analysis of the slope of 1/serum creatinine vs.time plots in these patients indicates no significant change in the slope after the initiation of epogen therapy.
Zidovudine-treated HIV-infected patients
Hypertension:Exacerbation of hyertension has not been observed in zidovudine-treated HIV-infected patients treated with epogen.However,epogen should be withheld in these patients if pre-existing hypertension is uncontrolled,and should not be started until blood pressure is controlled.In double-blind studies,a single seizure has been experienced by an epogen-treated patient.
Cancer patients on chemotherapy
Hypertension:Hypertension,associated with a significant increase in hematocrit,has been noted rarely in epogen-trated cancer patients.Nevertheless,blood pressure in epogentreated patients should be monitored carefully,particularly in patients with an underlying history of hypertension or cardiovascular disease.
Seizures:In double-blind,placebo-controlled trials,3.2%(N=2/63)of epogen-treated patients and 2.9%(N=2/68)of placebo-treated patients had seizures.Seizures in 1.6%(N=1/63)of epogen-treated patients occurred in the context of a significant increase in blood pressure and hematocrit from baseline values.However,both epogen-treated patients also had underlying CNS pathology which may have been related to seizure activity.
Thrombotic events:In double-blind,placebo-controlled trials,3.2%(N=2/63)of e pogen-treated patients and 11.8%(N=8/68)of placebo-treated patients had thrombotic events(e.g.,pulmonary embolism,cerebrovascular accident).
Growth Factor Potential:epogen is a growth factor that primarily stimulates red cell production.However,the possibility that epogen can act as a growth factor for any tumor type,particularly myeloid malignancies,cannot be excluded.
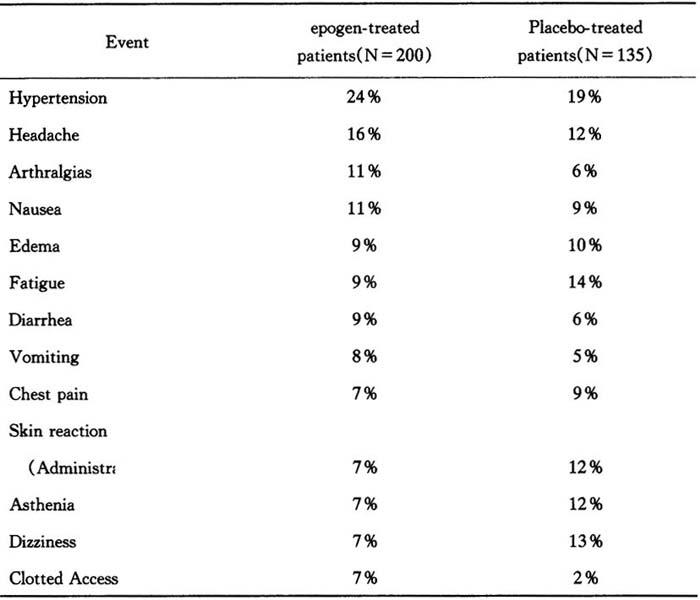
Adverse reactions
Chronic renal failure patients
Studies analyzed to date indicate that epogen is generally well-tolerated.The adverse events reported are frequent sequelae of CRF and are not necessarily attributable to epogen therapy.In double-blind,placebo-controlled studies involving over 300 patients with CRF,the events reported in greater than 5% of epogen-treated patients during the blinded phase were:
Table 9 Percent of patients reporting event
Significant adverse events of concern in patients with CRF treated in double-blind,placebo-controlled trials occurred in the following percent of patients during the blinded phase of the studies:
Seizure 1.1% 1.1%
CVA/TIA 0.4% 0.6%
MI 0.4% 1.1%
Death 0.0% 1.7%
In the U.S.epogen studies in patients on dialysis(over 567 patients),the incidence(number of events per patient-year)of the most frequently reported adverse events were:hypertension(0.75),headache(0.40),tachycardia(0.31),nausea/vomiting(0.26),clotted vascular access(0.25),shortness of breath(0.14),hyperkalemia(0.11),and diarrhea(0.11).Other reported events occurred at a rate of less than 0.10 events per patient per year.
Events reported to have occurred within several hours of administration of epogen were rare,mild,and transient,and included flu-like symptoms such as arthralgias and myalgias.
In all studies analyzed to date,epogen administration was generally well-tolerated,irrespective of the route of administration.
Allergic reactions:There have been no reports of serious allergic reactions or anaphylaxis associated with epogen administration.Skin rashes and urticaria have been observed rarely and when reported have been mild and transient in nature.There has been no evidence for development of antibodies to erythropoietin in patients tested to date,including those receiving intravenous epogen for over two years.Nevertheless,if an anaphylactoid reaction occurs,epogen should be immediately discontinued and appropriate therapy initiated.
Seizures:The relationship,if any,of epogen therapy to seizures is uncertain.The baseline incidence of seizures in the untreated dialysis population is difficult to determine;it appears to be in the range of 5-10% per patient year.There have been 47 seizures in 1 010 patients on dialysis,treated with epogen with an exposure of 986 patient-years,for a rate of approximately 0.048 events per patient-year.However,there appeared to be a higher rate of seizures during the first 90 days of therapy(occurring in approximately 2.5% of patients),when compared to subsequent 90-day time periods.WhiIe the relationship between seizures and the rate of rise of hematocrit is uncertain,it is recommended that the dose of epogen be decreased if the hematocrit increase exceeds 4 points in any 2-week period.
Hypertension:Up to 80%of patients with CRF have a history of hypertension.Blood pressure may rise during epogen therapy in CRF patients whether or not maintained on dialysis;during the early phase of treatment when hematocrit is increasing,approximately 25% of patients on dialysis may require initiation or increases in antihypertensive therapy.Hypertensive encephalopathy and seizures have been observed in patients with CRF treated with epogen.Increases in blood pressure may be associated with the rate of increase in hematocrit.It is recommended that the dose of epogen be decreased if the hematocrit increase exceeds 4points in any 2-week period.
Increases in blood pressure have been reported in clinical trials,often during the first 90 days of therapy.When data from all patients in the U.S.PhaseⅢmulticenter trial were analyzed,there was an apparent trend of more reports of hypertensive adverse events in patients on dialysis with a faster rate of rise of hematocrit(greater than 4 hematocrit points in any two week period).However,in a double-blind,placebo-controlled trial,hypertensive adverse events were not reported at an increased rate in the epogen-treated group(150 U/kg T.I.W.)relative to the placebo group.There do not appear to be any direct pressor effects of epogen.Special care should be taken to closely monitor and control blood pressure in epogen-treated patients.
Thrombotic events:During hemodialysis,patients treated with epogen may require increased anticagulation with heparin to prevent clotting of the artificial kidney.Clotting of the vascular access has occurred at an anualized rate of about 0.25 events per patient-year on epogen therapy.
A relationship has not been established with statistical certainty between a rise in hematocrit and the rate of thrombotic events[including thrombosis of vascular access(A-V shunt)]in epogen-treated patients.Overall,for patients with CRF(whether on dialysis ornot),other thrombotic events(e.g.,myocardial infarction,cerebrovascular accident,transient ischemic attack)have occurred at an annualized rate of less than 0.04 events per patient-year of epogen therapy.Patients with pre-existing vascular disease should be monitored closely.
Zidovudine-Treated HIV-infected patients
Adverse events reported in clinical trials with epogen in zidovudine-treated HIV-infected patients were consistent with the progression of HIV infection.In double-blind,placebocontrolled studies of 3-months duration involving approximately 300 zidovudine-treated HIVinfected patients,adverse events with an incidence of≥10%in either epogen-treated patients or placebo-treated patients were:
Table 10 Percent of patients reporting event
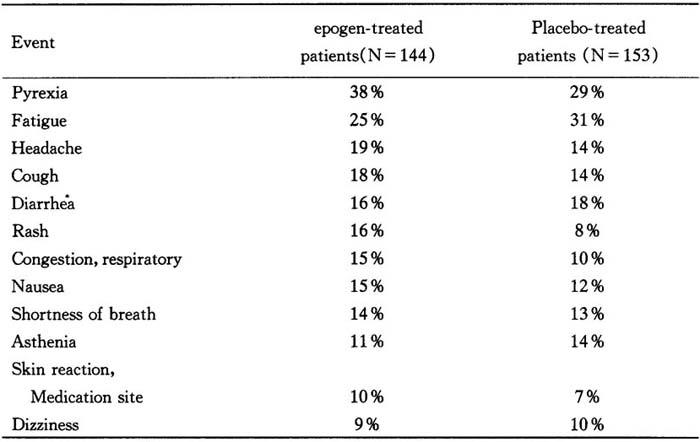
There were no statistically significant differences between treatment groups in the incidence of the above events.
In the 297 patients studied,epogen was not associated with significant increases in opportunistic infections or mortality.In 71 patients from this group treated with epogen at 150 U/kg T.I.W.,serum p24 antigen levels did not appear to increase.Preliminary data showed no enhancement of HIV replication in infected cell lines in vitro.
Peripheral white blood cell and platelet counts are unchanged following epogen therapy.
Allergic reactions:Two zidovudine-treated HIV-infected patients had urticarial reactions within 48 hours of their first exposure to study medication.One patient was treated with epogen and one was treated with placebo(epogen vehicle alone).Both patients had positive immediate skin tests against their study medication with a negative saline control.The basis for this apparent pre-existing hypersensitivity to components of the epogen formulation is un..known,but may be related to HIV-induced immunosuppression or prior exposure to blood products.
Seizures:In double-blind and open label trials of epogen in zidovudine-treated HlV-infected patients,10 patients have experienced seizures.In general,these seizures appear to be related to underlying pathology such as meningitis or cerebral neoplasms,not epogen therapy.
Cancer patients on chemotherapy
Adverse experiences reported in clinical trials with epogen in cancer patients were consistent with the underlying disease state.In double-blind,placebo-controlled studies of up to 3 months duration involving 131 cancer patients on chemotherapy,adverse events with an incidence>10%in either epogen-treated or placebo-treated patients were as indicated below:
Table 11 Percent of patients reporting event
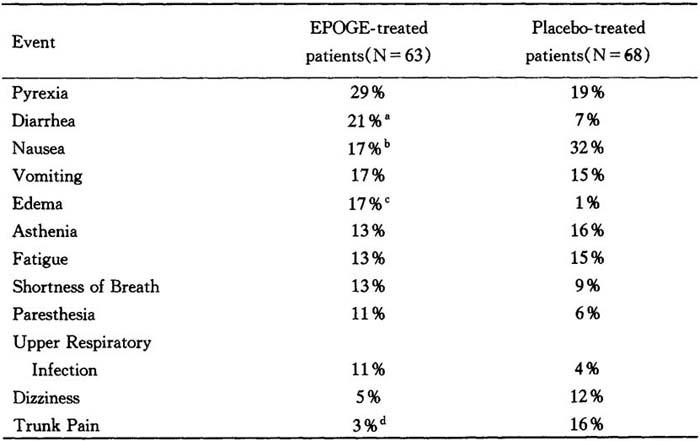
a:p=0.041.b:p=0.069.c:p=0.0016.d:p=0.017
Although some statistically significant differences between epogen-and placedo-treated patients were noted,the overall safety profile of epogen appeared to be consistent with the disease process of advanced cancer.During double-blind and subsequent openlabel therapy in which patients(N=72 for total epogen exposure)were treated for up to 32 weeks with doses as high as 927 U/kg,the adverse experience profile of epogen was consistent with the progression of advanced cancer.
Based on comparable survival data,and on the percentage of epogen-and placebo-treated patients who discontinued therapy due to death,disease progression,or adverse experiences (22%and 13%,respectively;p=0.25),the clinical outcome in the epogen-and placebotreated patients appeared to be similar.Available data from animal tumor models and measurement of proliferation of solid tumor cells from clinical biopsy specimens in response to epogen suggest that epogen may potentiate tumor growth.Nevertheless,as a growth factor,the possibility that epogen may not potentiate growth of some tumors,particularly myeloid tumors,cannot be excluded.A randomized controlled Phase IV study is currently ongoing to further evaluate this issue.The mean peripheral white blood cell count was unchanged following epogen therapy compared to the corresponding value in the placebo-treated group.
Overdosage
The maximum amount of epogen that can be safely administered in single or multiple doses has not been determined.Doses of up to 1 500 U/kg T.I.W.for 3-4 weeks have been administered without any direct toxic effects of epogen itself..
Therapy with epogen can result in polycythemia if the hematocrit is not carefully monitored and the dose appropriately adjusted.If the target range is exceeded,epogen may be temporarily withheld until the hematocrit returns to the target range;epogen therapy may then be resumed using a lower dose(see”Dosage and Administration”).If polycythemia is of concern,phlebotomy may be indicated to decrease the hematocrit.
Dosage and administration
Chronic renal failure patients
Starting doses of epogen over the range of 50-100 U/kg three times weekly(T.I.W.)have been shown to be safe and effective in increasing hematocrit and eliminating transfusion dependency in patients with CRF(see“Clinical Experience”).The dose of epogen should be reduced when the hematocrit reaches the target range of 30%-33%or increases by more than 4 points in any 2-week peiod.The dosage of epogen must be individualized to maintain the hematocrit within the target range.Dose changes should generally be in the range of 25 U/kg,T.I.W.The table below provides general therapeutic guidelines.
Starting dose: 50-100 U/kg T.I.W.;IV:Dialysis Patients IV or SC:Non-dialysis CRF patients
Reduce dose When:Target range is reached,or Hct.increases>4 points in any 2-week period
Increase dose If:Hct.does not increase by 5-6 points after 8 weeks of therapy,and hct.is below target range.
Maintenance dose: Individually titrate
Target Hct.range: 30%-33%(max.36%)
In patients on dialysis,epogen usually has been administered as an IV bolus T.I.W.While the administration of epogen is independent of the dialysis procedure,epogen may beadministered into the venous line at the end of the dialysis procedure to obviate the need for additional venous access.In patients with CRF not on dialysis,epogen may be given either as an intravenous or subcutaneous injection.
During therapy,hematological parameters should be monitored regularly(see“Laboratory Monitoring”).
Pre-therapy iron evaluation:Prior to and during epogen therapy,the patient’s iron stores,including transferrin saturation(serum iron divided by iron binding capacity)and serum ferritin,should be evaluated.Transferrin saturation should be at least 20%,and ferritin should be at least 100 ng/mL.Supplemental iron may be required to increase and maintain transferrin saturaiton to levels that will adequately support epogen-stimulated erythropoiesis.
Dose adjustment
When the hematocrit reaches 30%-33%,the dosage should be decreased by approximately 25 U/kg T.I.W.,to avoid exceeding the target range.One the hematocrit is within the target range,the maintenance dose must be individualized for each patient(see“Maintenance Dose”).
At any time,if the hematocrit increases by more than 4 points in a 2-week period,the dose should be immediately decreased.After the dose reduction,the hematocrit should be monitored twice weekly for 2-6 weeks,and further dose adjustments should be made as outlined in“Maintenance Dose.”
As the hematocrit approaches,or if it exceeds 36%,epogen should be temporarily withheld until the hematocrit decreases to the target range of 30%-33%;the dose should be reduced by approximately 25 U/kg T.I.W.upon re-initiation of therapy.
If a hematocrit increase of 5-6 points is not achieved after an eight week period and iron stores are adequate(see“Delayed or Diminished Response”),the dose of epogen may be increased in increments of 25 U/kg T.I.W.Further increases of 25 U/kg T.I.W.may be made at 4-6 week intervals until the desired response is attained.
Maintenance dose:The maintenance dose must be individualized for each patient.As the hematocrit approaches,or if it exceeds,36%,epogen should be temporarily with-held until the hematocrit is 33%or less.Upon re-initiation of therapy,the dose should be reduced by approximately 25 U/kg T.I.W.,or doses omitted,and an appropriate time interval(i.e.,2-6 weeks)allowed for stabilization of response.
If the hematocrit remains below,or falls below,the target range,iron stores should be reevaluated.If the transferrin saturation is less than 20%,supplemental iron should be administered.If the transferrin saturation is greater than 20%,the dose of epogen may be increased by 25 U/kg T,I.W.Such dose increases should not be made more frequently than once a month,unless clinically indicated,as the response time of the hematocrit to a dose increase can be 2-6 weeks.Hematocrit should be measured twice weekly for 2-6 weeks following dose increases.
In the U.S.Phase Ⅲ multicenter trial in patients on hemodialysis,the median maintenance dose was 75 U/kg T.I.W.,with approximately 65%of the patients requiring doses of 100 U/kg T.I.W.,or less,to maintain their hematocrit within the range of 32%-38%(maintenance doses ranged from 12.5-525 U/kg T.I.W.).Almost 10%of the patients required a dose of 25 U/kg,or less,and approximately 10%of the patients required more than 200 U/kg T.I.W.to maintain their hematocrit in this range.
In patients with CRF not on dialysis,the maintenance dose must also be individualized.Epogen doses of 75-150 U/kg per week have been shown to maintain hematocrits of 36%-38%for up to six months.
Delayed or diminished response:Over 95%of patients with CRF responded with clinically significant increases in hematocrit,and virtually all patients were transfusion-independent within approximately 2 months of initiation of epogen therapy.
If a patient fails to respond or maintain a response,other etiologies should be considered and evaluated as clinically indicated.See“Precautions”section for discussion of delayed or diminished response.
Zidovudine-treated HIV-infected patients
Prior to beginning epogen,it is recommended that the endogenous serum erythropoietin level be determined(pror to transfusion).Available evidence suggests that patients receiving zidovudine with endogenous serum erythropoietin levels>500 U/l are unlikely to respond to therapy with epogen.
Starting dose:For patients with serum erythropoietin levels≤500 U/l who are receiving a dose of zidovudine≤4 200 mg/week,the recommended starting dose of epogen is 100 U/kg as an intravenous or subcutaneous injection three times weekly(T.I.W.)for 8 weeks.
Increase dose:During the dose adjustment phase of therapy,the hematocrit should bemonitored weekly.If the response is is not satisfactory in terms of reducing transfusions requirements or increasing hematocrit after 8 weeks of therapy,the dose of epogen can be increased by 50-100 U/kg T.I.W.Response should be evaluated every 4-8 weeks thereafter and the dose adjusted according by 50-100 U/kg increments T.I.W.If patients have not responded satisfactorily to an epogen dose of 300 U/kg T.I.W.,it is unlikely that they will respond to higher doses of epogen.
Maintenance dose:After attainment of the desired response(i.e.,reduced transfusion requirements or increased hematocrit),the dose of epogen should be titrated to maintain the response based on factors such as variations in zidovudine dose and the presence of intercurrent infectious or inflammatoy episodes.If the hematocrit exceeds 40%,the dose should be discontinued until the hematocrit drops to 36%.The dose should be reduced by 25%when treatment is resumed and then titrated to maintain the desired hematocrit.
Cancer patients on chemotherapy
Baseline endogenous serum erythropoietin levels varied among patients in these trials with approximately 75%(N=83/1 10)having endogenous serum erythropoietin levels<132 U/l,and approximately 4 percent(N=4/1 10)of patients having endogenous serum erythropoietin levels>500 U/l.In general,patients with lower baseline serum erythropoietin levels responded more vigorously to epogen than patients with higher erythropoietin levels.Although no specific serum erythropoietin level can be stipulated above which patients would be unlikely to respond to epogen therapy,treatment of patients with grossly elevated serum erythropoietin levels(e.g.,>200 U/l)is not recommended.The hematocrit should be monitored on a weekly basis in patients receiving epogen therapy until hematocrit becomes stable.
Starting dose:The recommended starting dose of epogen is 150 U/kg subcuta-neously T.I.W.
Dose adjustment:If the response is not satisfactory in terms of reducing transfusions requirements or increasing hematocrit after 8 weeks of therapy,the dose of epogen can be increased up to 300 U/kg T.I.W.If patients have not responded satisfactorily to and epogen dose of 300 U/kg T.I.W.,it is unlikely that they will respond to higher doses of epogen.If the hematocrit exceeds 40%,the dose of epogen should be withheld until the hematocrit falls to 36%.The dose of epogen should be reduced by 25%when treatment is resumed and titrated to maintain the desired hematocrit.If the initial dose of epogen includes a very rapid hematocrit response(e.g.,an increase of more than 4 percentage points in any 2-week period),the dose of epogen should be reduced.
Preparation and administration of epogen
1.Do not shake.Shaking may denature the glycoprotein,rendering it biologically inactive.
2.Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration.Do not use any vials exhibiting particulate matter or discoloration.
3.Using aseptic techniques,attch a sterileneedle to a Sterile syringe.Remove the flip top from the vial Contaning epogen,and wipe the septum with a disinfectant.Insert the needle into the vial,and withdraw into the syringe an appropriate volume of solution.
4.Use only one dose per vial;do not re-enter the vial.Discard unused portions.Contains no preservative.
5.Do not administer in conjunction with other drug solutions.
How supplied
Epogen is available in vials containing 2 000(NDC 55513-126-01),3 000(NDC 55513-267-01),4 000(NDC 55513-148-01),or 10 000(NDC 55513-144-01),units of Epoetin alfa in 1.0 ml of a sterile,preservative-free solution.Each dosage form is supplied in boxes containing 10 single-use vials.
Storage
Store at 2-8℃.Do not freeze or shake.
Manufacturer
Amgen Inc.
Amgen Center
Thousand Oaks,California 91320-1789,U.S.A.
*生血素
〔活性成分〕
重组人红细胞生成素-β
〔性状〕
冻干粉,溶解后可作皮下注射或静脉注射。
〔成分〕
生血素1000:每瓶含1000U红细胞生成素-β冻干粉,相当于8.3μg促红素-β,每瓶附一安瓿1ml注射用水;
生血素2000:每瓶含2000U红细胞生成素-β冻干粉,相当于16.6μg促红素-β,每瓶附一安瓿1m1注射用水;
生血素5000:每瓶含5000U红细胞生成素-β冻干粉,相当于41.5μg促红素-β,每瓶附一安瓿1ml注射用水;
生血素10000:每瓶含10000U红细胞生成素-β冻干粉,相当于83μg促红素-β,每瓶附一安瓿1ml注射用水。
〔适应证〕
生血素适用于因慢性肾衰竭引致贫血的透析病人。生血素适用于治疗未行透析慢性肾功能不全病者的症状性肾性贫血。
〔禁忌证〕
生血素禁用于高血压失控的病人及对此药过敏的病人。
〔妊娠和哺乳期〕
虽然动物测试显示生血素在治疗条件下无致畸效应,但在妊娠和哺乳期不主张使用生血素,因为现时在这方面的临床经验仍不足。
〔药物不良反应〕
心血管系统:在治疗期间出现的最常见不良反应为血压升高或高血压情况恶化,特别是血细胞比容迅速增高的病例。这些都能以适当药物治疗。如血压升高不能以药物控制,须暂时停用生血素。病人除应定时检测血压,还应在各次透析之间进行检测(尤其在治疗初期)。
伴有脑病样症状(如头痛、精神混乱、感觉中枢和/或运动中枢紊乱,例如谈话障碍、步态失调和阵挛性癫痫发作等)的高血压危象可能会在正常血压或低血压病人身上出现。对这类病人应立即予以密切检测,特别是出现急性针刺性偏头痛者。
血液:生血素可能导致血小板数量在正常范围内轻微上升,特别是经静脉注射,情况一般随继续治疗消失。虽然血小板增多症极为罕见,但亦建议在治疗最初8周时,定期检测血小板量。
使用生血素的血液透析病人,往往因红细胞压积升高而需增加肝素剂量。如肝素剂量不适当,可能会发生透析器堵塞。偶尔会出现分流处栓塞,特别是有低血压趋向或动静脉瘘呈现并发症(血管狭窄、动脉瘤)的病人,所以建议及早预防血栓塞发生(如使用阿斯匹林)。
大多数病例中治疗期间同时出现血清铁蛋白下降及红血球压积升高。所以如病人血清铁蛋白低于100ng/ml或转铁蛋白饱和度低于20%,需每天口服补充铁质200~300mg。在个别病例中,病人可能出现短暂性高钾血症,高磷酸盐血症,所以治疗期间应定期检测。
其他:个别病人曾出现类似过敏反应。
〔警告〕
正常人如错误使用(例如用作刺激药物),可能会导致血细胞比容过多,因而引起各种致命的心血管系统并发症。
〔注意〕
小儿使用生血素还未积累足够的经验,2岁以下幼童暂不宜使用生血素。有恶性肿瘤、癫痫症、血小板增多、慢性肝衰竭的病人,使用生血素应当谨慎。
叶酸或维生素B12不足,会降低生血素的疗效。严重铝过多也会影响生血素的疗效。
对未行透析肾硬化病来说,使用生血素应看个体而定,因为促进肾衰竭加速的可能性不能排除。血清钾及磷酸盐必须定期检测,曾有极少数尿毒症病人在使用生血素后出现高钾情况,但其因果关系并不能确定。如发现高钾情况,应考虑停止使用生血素至钾回复正常水平止。
〔相互作用〕
到目前为止,临床结果还未显示生血素与其他药物有任何相互作用。
〔剂量〕
治疗目标为增加血细胞比容在30%~35%之间,每周平均增幅最少0.5%,最高水平不能超过35%。如存在高血压或心血管疾病,脑血管或末梢血管病症等情况,应按个体状况来确定每周升幅及目标水平。对部分病人来说,血细胞比容理想水平可能低于30%。
生血素治疗方案分两步骤:
1.治疗期
皮下注射:开始时治疗剂量为每周3×20U/kg。如发现血细胞比容增加不理想(每周增加少于0.5%),可于每4周后每周剂量增加3×20U/kg。也可将每周剂量分成每日剂量。
静脉注射:开始时治疗剂量为每周3×40U/kg。如发现血细胞比容增加不理想(每周增加少于0.5%),可于4周后每周剂量增加至3×80U/kg。其后每间隔1个月每周可再增多3×20U/kg。
以上两种给药途径,最高剂量不可超过每周720U/kg。
2.维持期 要达到维持血细胞比容在30%~35%之间,首先把剂量减至治疗期剂量的二半,然后每周或每2周调整剂量。
对儿童所做临床观察结果显示,平均来说,病人越年青,生血素维持剂量越高,然而,由于个别病人反应不同,难于估计,所以应按照建议剂量应用本药。
〔疗程〕
生血素一般用于终身治疗,但如有需要,可以随时终止疗程。
〔用法〕
生血素应在医务人员仔细监护及指导下使用。
由于曾发现个别过敏性反应病例,建议首次给药应在医务人员监护下进行。
生血素是以冻干粉存于小瓶内供应。
使用时将生血素溶解于附带的溶剂,但应马上使用(2h之内)。在使用前,确定溶液应呈无色无颗粒状态,透明或呈轻微乳状,方可使用。用剩药物应丢弃。
本药溶液可用于皮下注射或静脉注射。
静脉注射应在大约2min内完成。例如,在透析结束时经动静脉瘘管注入。非透析病人应采用皮下注射以避免刺伤外周静脉。
〔重要配伍禁忌〕
注意以下提示,避免引致不相配伍或影响活性:
1.切勿使用其他溶剂!
2.切勿与其他药物混合使用!
3.切勿使用玻璃注射用具,应使用塑料制品!
〔贮存〕
生血素应贮存于2~8℃(冷藏箱)。
运输时,冷藏温度如遭间断,时间不可超过5d及温度一定不超过25℃。
〔有效期〕
生血素必须在印于盒上的有效期内使用。
〔包装〕
生血素1000,生血素2000:每盒10瓶冻干粉和10安瓿注射溶剂。
生血素5000,生血素10000:每盒5瓶冻干粉和5安瓿注射溶剂。
切勿让儿童触及!
OPC-安瓿开启方法:不需使用锉刀!
如何开启新设计OPC安瓿:
1.手持安瓿,保持蓝点向上位置。振摇或叩击安瓿使溶剂不留于安瓿茎部。
2.手持安瓿,保持蓝点向上位置。拿稳茎部,向外折断。
〔生产厂家〕
德国Boehringer mannheim GmbH
*Recormon
Active ingredient
Epoetin beta.(Synonyms:rhEPO and recombinant humanerythropoietin).
Presentation
Freeze-dried substance for subcutaneous or intravenous administration after dissolution.
Composition
Recormon 1 000:1 vial contains 1 000 international units epoetin beta as a freeze-dried substance,corresponding to 8.3 μg epoetin beta.
1 solvent ampoule contains 1 ml water for injections.
Recormon 2 000:1 vial contains 2 000 international units epoetin beta as a freeze-dried substance,corresponding to 16.6μg epoetin beta.
1 solvent ampoule contains 1 ml water for injections.
Recormon 5 000:1 vial contains 5 000 international units epoetin beta as a freeze-dried substance,corresponding to 41.5μg epoetin beta.
1 solvent ampoule contains 1 ml water for iniections.
Recormon 10 000:1 vial contains 10 000 international units epoetin beta as a freezedried substance,corresponding to 83 μg epoetin beta.
1 solvent ampoule contains 1 ml water for injections.
Indication
Recormon is used for the treatment of anaemia associated with chronic renal failure in patients on dialysis.Recormon is used for the treatment of symptomatic renal anaemia in patients with chronic renal insufficiency not yet undergoing dialysis.
Contraindications
Recormon must not be used in poorly controllable hypertension and known hypersensitivity to the medication.
Use in pregnancy and lactation period
Animal studies revealed that no teratogenic effects occur under therapeutic conditions.However,Recormon should not be used during pregnancy and lactation as at present insufficient clinical experience has been gained in these fields.
Adverse drug reactions
Cardiovascular system:the most frequent adverse reaction during treatment with Recormon is an increase in blood pressure or aggravation of existing hypertension,especially in cases of rapid PCV increase.These increases in blood pressure can be treated with drugs.If blood pressure rises cannot be controlled by drug therapy,a transient interruption of Recormon therapy is recommended.Regular monitoring of blood pressure is recommended,also between dialyses(particularly at the beginning of therapy).Hypertensive crisis with encephalopathy-like symptoms(e.g.headache,confusion,sensorimotor disorderssuch as speech disturbance,impaired gait-up to tonoclonic seizures)may occur,also in individual patients with normal or low blood pressure.This requires the immediate attention of a physician and intensive medical care.Particular attention should be paid to sudden stabbing migraine-like headaches as a possible warning signal.
Blood:There may be a moderate dose-dependent rise in the platelet count within the normal range during treatment with Recormon,especially after intravenous administration.This regresses during the course of continued therapy.Development of thrombocytosis is very rare.It is recommended that the platelet count is regularly monitored during the first 8 weeks of therapy.An increase in heparin dose during haemodialysis is frequently required during the course of therapy with Recormon as a result of the increased packed cell volume.Occlusion of the dialysis system is possible if heparinization is not optimum.Shunt thromboses may occur,especially in patients who have a tendency to hypotension or whose arteriovenous fistulae exhibit complications(e.g.stenoses,aneurysms).Early shunt revision and thrombosis prophylaxis,e.g.with acetylsalicylic acid,is therefore recommended in these patients.
In most cases,a fall in serum ferritin values simultaneous with a rise in packed cell volume is observed.Therefore oral iron substitution with 200 to 300 mg Fe2+/day is recommended in all patients with serum ferritin values below 100 ng/ml or transferrin saturation below 20%.In isolated cases,transient hyperkalaemia and hyperphosphataemia may occur.These parameters should be monitored regularly.
Others:Anaphylactoid reactions have been observed in isolated cases.
Warning
Misuse by healthy persons(e.g.for doping)may lead to an excessive increase in packed cell volume.This may be associated with life-threatening complications of the cardiovascular system.
Special precautions for use
Recormon should not be used in children below the age of 2 years as at present insufficient clinical experience has been gained.Recormon should be used with caution in the presence of malignant tumours,epilepsy,thrombocytosis and chronic liver failure.Folic acid and vitamin B12 deficiencies should be ruled out as they reduce the effectiveness of Recormon.Severe aluminium overload may compromise the effectiveness of Recormon.The indication for Recormon treatment of nephrosclerotic patients not yet undergoing dialysis should be defined individually as a possible acceleration of progression of renal failure cannot be ruled out with certainty.Serum potassium and phosphate levels should be monitored regularly during Recormon therapy.Potassium elevation has been reported in a few uraemic patients receiving Recormon,though causality has not been established.If an elevated or rising potassium level is observed,then consideration should be given to ceasing Recormon administration until the level has been corrcted.
Interaction
The clinical results obtained so far do not indicate any interaction of Recormon with other substances.
Dosage(single and weekly dose)
The aim of treatment is to increase the packed cell volume to 30%-35%whereby the weekly increase should be at least 0.5%.A value of 35%should not be exceeded.
In the presence of hypertension or existing cardiovascular,cerebrovascular or peripheral vascular diseases,the weekly increase in PCV and the target PCV should be determined individually taking into account the clinical picture.In some patients the optimum PCV may be below 30%.Treatment with Recormon is divided into two stages:
Correction phase:
·Subcutaneous administration
The initial dosage is 3×20 U/kg body weight and week.The dosage may be increased every 4 weeks by 3× 20 U/kg body weight and week if the increase of packed cell volume is not adequate(less than 0.5%per week).The weekly dose can also be divided into daily doses.
·Intravenous administration
The inital dosage is 3 × 40 U/kg body weight and week.The dosage may be raised after 4 weeks to 3×80 U/kg body weight and week if the increase of packed cell volume is not adequate(less than 0.5%per week).Further increments of 3 × 20 U/kg body weight and week are possible at monthly intervals.
For both routes of administration,the maximum dosage should not exceed 720 U/kg body weight and week.
·Maintenance phase:
To maintain a packed cell volume of between 30%-35%,the dosage is initially reduced to half of the previously administered amount.Subsequently,the dose is adujsted at intervals of one or two weeks individually for the patient(maintenance dose).
Results of clinical studies in children have shown that,on average,the younger the pa tients,the higher the Recormon doses.Nevertheless,the recommended dosing schedule should be followed as the individual response cannot be predicted.
Duration of treatment
Treatment with Recormon is usually a life-long therapy.It can,however,be interrupted,if necessary at any time.
Method of administration
This product should only be used under consultant supervision,usually in a hospital setting.Since anaphylactoid reactionswere observed in isolated cases,it is recommended that the first dose be administered under medical supervision.
Recormon is supplied as a freeze-dried substance in vials.This is dissolved with the contents of the accompanying solvent ampoule.The reconstituted solution should be used immediately,i.e.within 2 hours.Only solutions which are clear or slightly opalescent,colourless and practically free of visible particles may be injected.Any medication remaining in the vial after use should be discarded.
The reconstituted solution can be administered subcutaneously or intravenously.
In case of intravenous administration,the solution should be injected over approx.2 minutes,e.g.in haemodialysis patients via the arterio-venous fistula at the end of dialysis.For non-haemodialyzed patients,subcutaneous administration should always be preferred in order to avoid puncture of peripheral veins.
Most important incompatibilities
To avoid incompatibility or loss of activity,the following instructions must be observed:
·Do not use any other solvent
·Do not mix with other drugs
·Do not use glass,but only plastic materials for injection
Instructions for storage
Recormon must be stored at a temperature of 2-8℃(refrigerator).For the purpose of transport,the cooling chain may be interrupted for a period of up to 5 days at room temperature(≤25℃).
Expiry date
Recormon should not be used after the expiry date printed on the pack.
Presentations and pack sizes
Recormon 1000,Recormon 2000:
10 vials with freeze-dried substance and 10 ampoules with solvent.
Recormon 5 000,Recormon 10 000:
5 vials with freeze-dried substance and 5 ampoules with solvent.
Keep all medicines out of the reach of children!
Note on usage of OPC(One-Point-Cut)ampoules:
Filing no longer necessary!How to use the new OPC ampoules.
Hold the ampoule with the blue point upwards.Shake or tap the ampoule to get any fluid in the stem into the body of the ampoule.
Hold the ampoule with the blue point upwards.Take hold of the stem and snap off away from you.
Manufacturer
Boehring Mannheim GmbH Germany